Article Text

Abstract
Objective Both excessive and insufficient activation of WNT signalling results in cartilage breakdown and osteoarthritis. WNT16 is upregulated in the articular cartilage following injury and in osteoarthritis. Here, we investigate the function of WNT16 in osteoarthritis and the downstream molecular mechanisms.
Methods Osteoarthritis was induced by destabilisation of the medial meniscus in wild-type and WNT16-deficient mice. Molecular mechanisms and downstream effects were studied in vitro and in vivo in primary cartilage progenitor cells and primary chondrocytes. The pathway downstream of WNT16 was studied in primary chondrocytes and using the axis duplication assay in Xenopus.
Results WNT16-deficient mice developed more severe osteoarthritis with reduced expression of lubricin and increased chondrocyte apoptosis. WNT16 supported the phenotype of cartilage superficial-zone progenitor cells and lubricin expression. Increased osteoarthritis in WNT16-deficient mice was associated with excessive activation of canonical WNT signalling. In vitro, high doses of WNT16 weakly activated canonical WNT signalling, but, in co-stimulation experiments, WNT16 reduced the capacity of WNT3a to activate the canonical WNT pathway. In vivo, WNT16 rescued the WNT8-induced primary axis duplication in Xenopus embryos.
Conclusions In osteoarthritis, WNT16 maintains a balanced canonical WNT signalling and prevents detrimental excessive activation, thereby supporting the homeostasis of progenitor cells.
- Chondrocytes
- Knee Osteoarthritis
- Osteoarthritis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Osteoarthritis (OA) is characterised by breakdown of the articular cartilage, abnormal bone remodelling, osteophytosis and, in some cases, low-degree synovitis. It is a leading cause of chronic disability, yet we still do not have a cure. Although generally efficacious, joint replacement does not live up to patient expectation in up to 30% of cases,1 it has a limited life span, often requiring complex revisions, and is not suitable for the ever-growing population of younger patients with early OA who have an active lifestyle.
WNT signalling is involved in several biological processes including joint formation during embryonic skeletogenesis and joint homeostasis and disease in postnatal life.2–11
WNTs are a family of 19 highly conserved morphogens that signal through several different pathways. In the β-catenin-dependent (or canonical WNT) pathway, engagement of WNTs with their receptors results in the accumulation and nuclear translocation of β-catenin and transcriptional activation of target genes.12 The non-canonical pathways are less understood and involve Ca2+ signalling, phosphatidylinositol-4,5-biphosphate 3-kinase (PI3K) and activation of cjun N-terminal kinase (JNK).13
Although genetic studies in humans and experimental studies in animals provide overwhelming evidence for the role of WNT signalling in cartilage homeostasis and OA, the mechanisms underpinning these functions are still poorly understood. It has been shown that a delicate balance of WNT activity is needed for cartilage homeostasis as both repression7 and constitutive activation8 of the β-catenin pathway lead to cartilage breakdown. Similarly, excessive WNT activation following loss of function of the WNT inhibitor Frizzled related protein (FRZB) resulted in increased susceptibility to OA in mice14 and in humans,15 but the excessive WNT suppression due to the tumour necrosis factor-dependent expression of DKK1 in inflammatory arthritis also results in cartilage and bone destruction.16 ,17 How this balance is achieved within the joint is unclear.
WNT16 is expressed in the joint interzones during joint formation and in the superficial cells of the articular cartilage postnatally.4 ,5 In adult cartilage, it is not expressed in normal conditions, but it is rapidly upregulated following injury or in OA.18
We demonstrate that WNT16 upregulation following injury protects cartilage from breakdown following destabilisation of the medial meniscus (DMM). This effect is likely mediated through upregulation of lubricin, an essential joint lubricant that protects chondrocytes from shear stresses and cartilage from mechanically induced breakdown.19 We also show that WNT16 is a weak activator of canonical WNT signalling in chondrocytes and that this is required for the upregulation of lubricin. However, in the presence of other more potent canonical WNT ligands, WNT16 acts as an inhibitor, preventing excessive activation of the pathway, which is known to result in cartilage damage.
Methods
Surgical procedure
DMM was performed as previously described20 ,21 on 10-week-old male wnt16−/− or wild-type 129/Sv mice (cousins). The contralateral knees were either sham-operated or non-operated as an internal control. The mice were sacrificed by cervical dislocation at 2 days, 7 days or 8 weeks post surgery.
Histological analysis and OA scoring
Histological processing was performed as discussed elsewhere21 and scored using the Osteoarthritis Research Society International (OARSI) scoring system by two independent investigators.22 All images were taken using the same settings on an Olympus BX61 microscope.
Immunohistochemistry and TUNEL staining
Immunohistochemistry and apoptosis detection were performed as described in refs 21, 23, 24 and detailed in supplementary materials.
Supplemental material
Gene expression analysis
DMM or control joints were detached from the animals immediately after death and the soft tissues removed under a dissecting microscope. The articular surface of the femoral condyles and the tibial plateaus was isolated at the level of the growth plate using a scalpel and homogenised in 1 mL of Trizol (Life Technologies).
In vitro gene expression analysis was performed on primary human or bovine articular chondrocytes (BACs) obtained from young animals (isolated as described in ref. 23) and on superficial zone cell (SZC) isolated from the knees of neonatal mice.5 Cells were stimulated with WNT16, WNT3a or DKK1 recombinant proteins (R&D Systems) for 24 h before phosphate buffered saline (PBS) wash and addition of 1 mL of Trizol (Invitrogen) for RNA isolation and Q-PCR analysis.23 A list of the primers used can be found in online supplementary table S1.
SUPER8XTOPFlash reporter assay
The chondrocytes were co-transfected with SUPER8XTOPFlash TCF/LEF-firefly luciferase reporter plasmid and CMV-Renilla luciferase plasmid using JetPRIME transfection reagent (Polyplus). The cells were stimulated for 48 h replacing the medium after 24 h before luciferase activity was measured using the Dual luciferase Reporter assay system (Promega).
Xenopus assay
Xenopus embryos were injected ventrally with either 0.02 ng/μL wnt16 or 0.002 ng/μL wnt8 RNA at the four-cell stage and left for 2 days to develop before analysis.
Statistical analysis
Parametric data were compared with the t test, non-parametric data with the Wilcoxon–Mann–Whitney test. For multiple comparisons, the analysis of variance or Kruskal–Wallis test, including the appropriate post test, was used. p Values <0.05 were considered significant: *p<0.05; **p<0.01; ***p<0.001.
Results
WNT16 is transiently upregulated in cartilage following DMM
We reported that the embryonic joint interzone marker WNT16 is re-expressed in the adult human articular cartilage following acute injury and in OA.18
We first confirmed that WNT16 was no longer expressed in healthy adult mouse articular cartilage, and that, following DMM, it was upregulated at mRNA and protein level (figure 1). Some upregulation also took place in sham-operated knees, particularly at early time points; however, DMM-operated knees exhibited a greater upregulation at 7 days.

WNT16 is upregulated in experimental osteoarthritis induced by destabilisation of the medial meniscus (DMM). Wild-type mice received DMM of the left knee and sham operation on the right and were killed at 2 days (C and D), 7 days (E and F) and 8 weeks (G) post surgery. (A) WNT16 mRNA was upregulated in the whole joints of DMM-operated mice compared with sham-operated controls, reaching statistical significance after 7 days. WNT16 protein was undetectable in non-operated knees (B), but was upregulated at 2 days following DMM (C) and, to a lesser extent, following sham operation (D). WNT16 expression was further increased in DMM-operated mice at 7 days (E) and to less of an extent following sham operation (F) (joint tissues labelled as follows: C, cartilage; B, subchondral bone; M, meniscus). After 8 weeks, (G) WNT16 was no longer detected in DMM-operated knees. (H) Isotype negative control at the 2-day time point. N=3 per condition.
WNT16-deficient mice have a more severe outcome following surgical induction of OA
To elucidate the role of WNT16 upregulation following joint injury, we generated WNT16 null mice (wnt16−/−) through deletion of exon 2 by homologous recombination (see online supplementary figure S1A). Successful recombination was confirmed by PCR (see online supplementary figure S1B). The mutants were backcrossed for nine generations in the 129/Sv genetic background where no differences were detected in cartilage morphology (see online supplementary figure S1C,D). At 18 months of age, both wild-type and wnt16−/− animals developed minor superficial cartilage irregularities in keeping with the strain. In the 129/Sv genetic background, we did not detect differences in bone structure/morphology as shown by Micro-CT at the vertebral bodies (see online supplementary figure S2) and the femoral epiphyses (see online supplementary figure S3). The thickness of the subchondral bone was also comparable (see online supplementary figure S3C). However, in keeping with a previous report,25 in the DBA/1 background, spontaneous fractures occurred in wnt16−/− mice (not shown), suggesting that the severity of the previously reported bone phenotype is strain dependent. These results suggest that WNT16 is not required for the normal development and growth of synovial joints and, in resting conditions, the absence of WNT16 does not predispose to OA.
Standard animal husbandry conditions are not always representative of normal life, where, in humans, cartilage injuries are common.26 Since WNT16 is upregulated in cartilage after injury18 in humans and mice, we investigated whether the upregulation of this molecule has a role in the adaptive responses that restore cartilage homeostasis following injury. To study this, we induced experimental OA in 10-week-old wnt16−/− and wild-type controls (129/Sv background) by DMM.20
Eight weeks following DMM, wnt16−/− mice developed more severe OA compared with wild-type controls as assessed by histological scoring22 ,27 (figure 2A, B) and histomorphometrical comparison of the intensity of safranin O staining (figure 2C). The histomorphometry is described in online supplementary figure S4. An independent experiment in mice at the fifth backcross into the 129/Sv phenotype yielded similar results (data not shown). We did not detect statistically significant differences in the expression of the aggrecanases ADAMTS-4/-5 or in the detection of aggrecan neoepitopes generated by aggrecanase or matrix metalloproteases (MMP) cleavage (see online supplementary figure S5).

Wnt16−/− mice are more susceptible to instability-induced osteoarthritis. The 10-week-old mice (7 wild-type (WT) and 10 wnt16−/− (KO)) were subjected to destabilisation of the medial meniscus and the outcome was compared 8 weeks following surgery. (A) Safranin O staining of the medial compartment of representative sections. (B) OARSI histological score. (C) Intensity of the safranin O in the medial tibial plateau. LF, lateral femur; LT, lateral tibia; MF, medial femur; MT, medial tibia.
These results suggest that WNT16 has a homeostatic role that limits the extent of cartilage breakdown following induction of joint instability.
WNT16-deficient mice fail to upregulate the joint lubricant lubricin, following surgical induction of OA
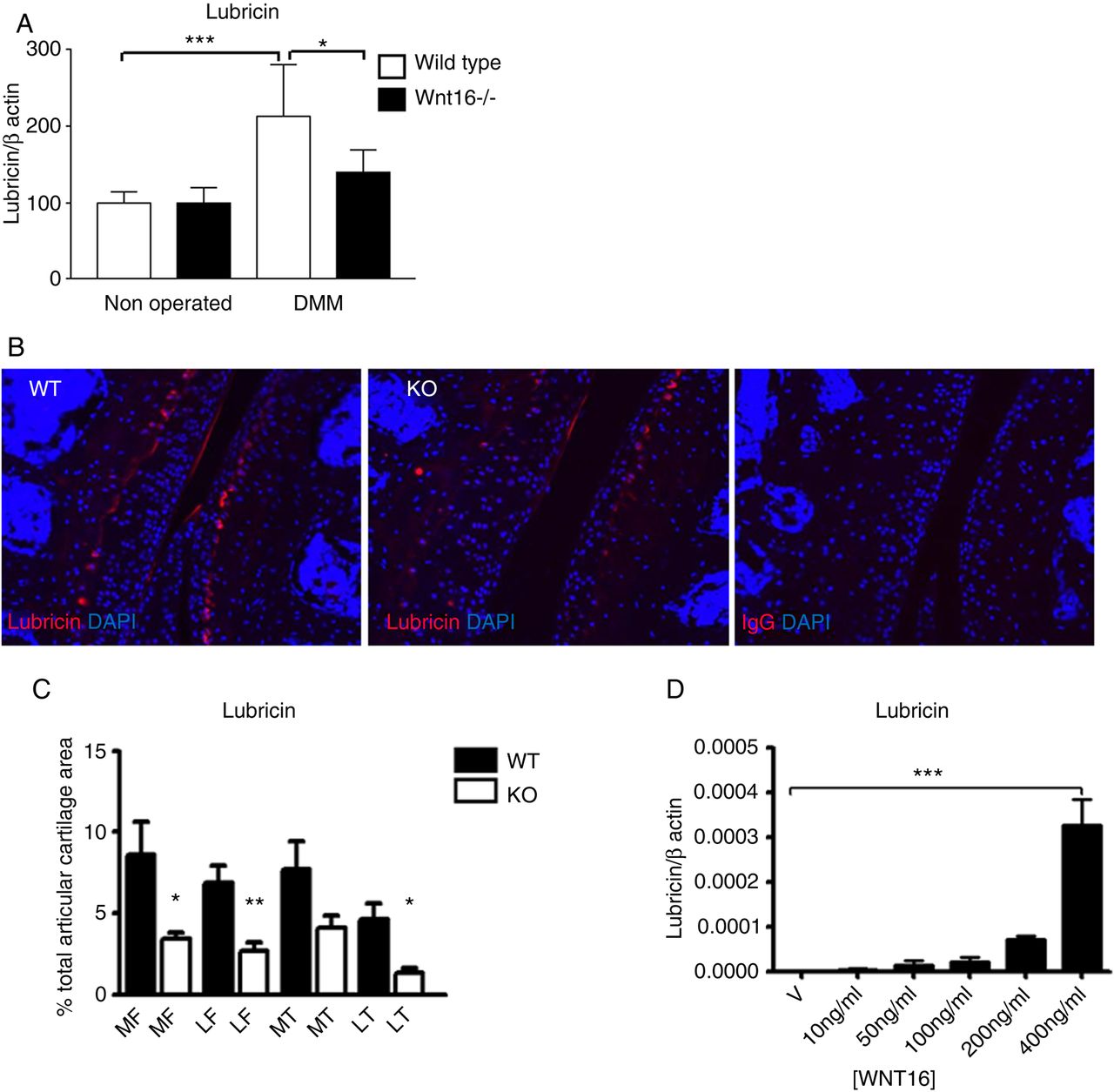
WNT signalling is essential for the expression of the joint lubricant lubricin.5 Deficiency of Prg4 (gene encoding for lubricin) in mice and loss of function mutations in humans leads to cartilage breakdown and spontaneous OA due to increased coefficient of friction in the joint.19 Wnt16−/− and wild-type mice showed similar levels of lubricin under resting conditions; however, 2 days following surgery, wild-type mice, but not wnt16−/−, upregulated lubricin expression at mRNA and protein level (figure 3A–C).

Lubricin was upregulated following destabilisation of the medial meniscus (DMM). Wild-type (WT) and wnt16−/− mice were subjected to DMM and killed 2 days thereafter. (A) The joint surface, including the articular cartilage and the subchondral bone, was dissected and processed for gene expression analysis by real-time PCR. Lubricin was upregulated in WT mice but not in wnt16−/− mutants at mRNA level 2 days following DMM (N≥10). (B) At protein level, lubricin staining following DMM was detected in the superficial layer of the articular cartilage and in a region between the intermediate and the deep layer in WT mice. (C) The area of the articular cartilage positive for lubricin staining 2 days following DMM was significantly higher in WT mice (N=4) than in wnt16−/− mice (N=6). (D) WNT16 recombinant protein upregulated lubricin mRNA expression in bovine articular chondrocyte (BAC) at 24 h (N=3). KO, knockout. *p<0.05; **p<0.01; ***p<0.001.
Treatment of primary BACs with recombinant WNT16 was sufficient to upregulate lubricin expression in vitro (figure 3D).
Loss of WNT16 resulted in increased apoptosis in the SZCs of the articular cartilage
Spontaneous OA development in Prg4-deficient mice is associated with chondrocyte apoptosis.19 Two days following DMM, when WNT16 is upregulated in wild-type mice, WNT16-deficient mice developed increased apoptosis. This selectively occurred in the superficial layer of the cartilage and, as expected, was particularly increased in the medial compartment of the knee where the osteoarthritic lesions are most severe (figure 4A, B).
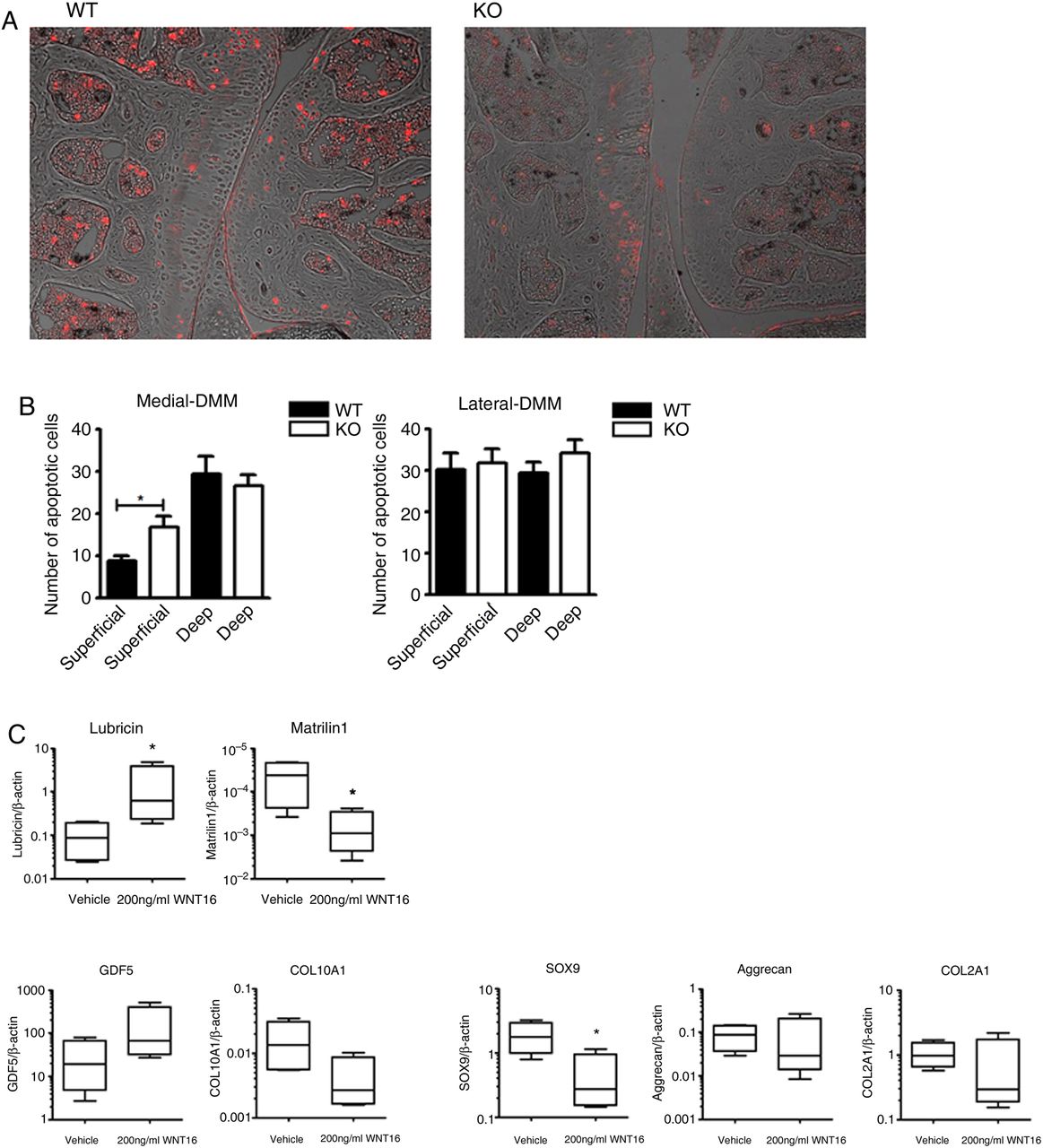
Disruption of WNT16 results in apoptosis of superficial cartilage cells and in the loss of superficial zone molecular markers. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining of representative sections from the medial compartment of the knee of wild-type (WT) and wnt16−/− mice 2 days after destabilisation of the medial meniscus (DMM) (N=6) (A). The TUNEL staining (shown in red) was superimposed on the Nomarski image for anatomical orientation. Quantification of TUNEL-positive cells in the superficial and deep layers of the articular cartilage within the medial compartment (most affected in DMM) and lateral compartment (mildly or unaffected) (B). Gene expression analysis of superficial zone cells from wnt16−/− mice with or without rescue with exogenous 200 ng/mL recombinant WNT16 (N=4) (C). KO, knockout. *p<0.05.
As the loss of WNT16 specifically affected apoptosis in the SZCs and as these cells specifically express WNT16 during embryonic and early postnatal development, we explored the effect of WNT16 in the SZC. In adulthood, these cells are considered a specific progenitor cell population within the permanent articular cartilage and are essential for cartilage homeostasis.28 Cells were isolated as previously described from the knees of neonatal mice.5 These cells display a marker profile characterised by expression of WNT16 and lubricin, and low expression of COL2A1, Aggrecan and Matrilin15 ,6 (see online supplementary figure S6). The remainder of the neonatal epiphysis is prevalently composed of epiphysial chondrocytes that are destined to be replaced by bone, do not express WNT16 and do express Matrilin1.29
In cultures enriched for SZC from wnt16−/− mice, recombinant WNT16 upregulated the SZC lineage marker lubricin, downregulated the epiphysial lineage marker Matrilin1 and inhibited the expression of SOX9, which is expressed at higher level in epiphysial chondrocytes compared with SZC (figure 4C).5 This suggested that rather than just upregulating lubricin, WNT16 specifically supports the phenotype marker profile of the SZC.
WNT16 maintains a balanced activation of the canonical pathway
Since canonical WNT signalling supports the phenotype of the superficial zone chondrocytes,5 ,6 we tested whether WNT16 could activate the canonical WNT pathway in chondrocytes and whether canonical WNT activation was required for lubricin upregulation by WNT16. In keeping with previous reports,3 ,25 WNT16 concentrations of 200 ng/mL or higher consistently and dose-dependently upregulated Axin2 (a transcriptional target of canonical signalling) in primary human and BACs (figure 5A and online supplementary figure S7A).

WNT16 is a weak activator of the β-catenin-dependent pathway but also limits its activation by other more potent canonical WNTs. WNT16 caused a weak upregulation of the canonical WNT reporter gene Axin2 compared with WNT3a, in human articular chondrocytes (HAC) (N=3 patients, 4 replicates each) (A). Lubricin expression caused by WNT16 stimulation (ng/mL) in HAC (B), and mouse superficial zone cell (SZC) (C), was higher than that caused by WNT3a stimulation. Lubricin expression decreased in the presence of canonical inhibitor DKK1 (100 ng/mL) in mouse SZC (N=4) (C). All at 24 h. Following destabilisation of the medial meniscus (DMM) surgery, wnt16−/− mice had a higher expression of Axin2 mRNA in their joints (D). WNT16 reduced the capacity of WNT3a to activate the SUPER8XTOPFlash reporter assay in HEK293 cells treated with WNT3a, alone and in combination with WNT16 (ng/mL) (N=4) (E and F). wnt16 inhibited axis duplication caused by wnt8 in Xenopus laevis (G and H). Embryos were injected ventrally with 0.02 ng/µL wnt16 or 0.002 ng/µL wnt8 at the four-cell stage and left for 2 days to develop (n=36–47).
Interestingly, although WNT16 was not as efficient as WNT3a at activating the canonical pathway, it caused a larger upregulation of lubricin (figure 5B, C and online supplementary figure S7B), which still was partially abolished by the canonical WNT inhibitor DKK1 (figure 5C and online supplementary figure S7C).
Since WNT16 could activate the canonical WNT pathway in vitro, we expected that wnt16−/− mice would display decreased Axin2 upregulation following experimental OA. Surprisingly, WNT16-deficient mice had higher levels of Axin2, indicating a higher level of canonical pathway activation (figure 5D). Since WNT16 is a weak activator of the canonical pathway (figure 5A), we hypothesised that, in vivo, WNT16 could compete with other, more potent, WNTs activated in OA,14 thereby limiting their capacity to disproportionately activate the β-catenin pathway, which is known to result in further cartilage breakdown. To confirm this hypothesis, we investigated whether WNT16 could stunt the WNT3a-induced activation of the canonical pathway. As expected, WNT3a alone dose-dependently activated the SUPER8XTOPFLASH reporter assay (figure 5E), but co-treatment with WNT16 dose-dependently inhibited the WNT3a-induced reporter activity starting from concentrations as low as 50 ng/mL (figure 5E, F).
To investigate whether WNT16 can inhibit the signalling of more potent canonical WNTs in vivo and in different biological contexts, we used the well-established Xenopus assay where injection of a ventral blastomere at the four-cell stage with WNT ligands that activate the canonical WNT pathway induces complete primary axis duplication or a strong hyperdorsalisation.30
As expected, injection of wnt8 induced complete axis duplication in the majority of the tadpoles, as defined by complete duplication of the cement gland (yellow arrowhead) (figure 5G, H). Consistent with our data, injection of wnt16 RNA mostly induced weak hyperdorsalisation, which indicates a very mild activation of the β-catenin pathway. However, co-injection of wnt16 and wnt8 resulted in inhibition of the complete axis duplication induced by wnt8 and in a phenotype resembling that induced by the injection of wnt16 alone (figure 5G, H).
We propose a model where WNT16 buffers the activation of the canonical WNT pathway, overcoming the activity of more potent canonical WNT ligands such as WNT3a and wnt8, and supporting a controlled mild activation of WNT signalling, while preventing the excessive activation mediated by other WNTs (figure 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cartilage injury causes activation of WNT signalling through downregulation of antagonists such as FRZB and upregulation of several agonists such as WNT16 and WNT8.14 ,18 WNT16 buffers the canonical WNT activation to homeostatic levels through its capacity to directly support a weak activation and preventing excessive activation induced by other ligands. Excessive canonical WNT activation causes cartilage breakdown by driving inappropriate maturation particularly within the superficial zone progenitor cells,8 ,18 whereas homeostatic levels of activation are necessary for supporting the superficial progenitor population and lubricin expression (this work and refs 5, 7).
Discussion
We discovered that, whereas WNT16 is dispensable during joint embryonic morphogenesis, when other Wnts (WNT9a and WNT4) are also expressed within the joint interzones, its upregulation following injury in adult life is essential to preserve cartilage homeostasis. This likely occurs through the unique capacity of this WNT ligand to prevent excessive activation of the canonical WNT pathway by limiting the capacity of other more potent WNT ligands and by contributing to the upregulation of lubricin, an essential joint lubricant that protects chondrocytes from shear stress. Through activation of the canonical WNT pathway, WNT16 was a specific activator of lubricin since WNT3a was more potent in activating the canonical WNT pathway but far less potent in upregulating lubricin.
Recently, it was reported that WNT16 produced by osteoblasts supports cortical bone formation and bone homeostasis by inhibiting osteoclastogenesis.25 Mutants lacking either exons 2 and 3 or the entire open-reading frame of wnt16 gene displayed the same phenotype.25 We originally backcrossed our mutants in the DBA/1 genetic background and also observed a high incidence of spontaneous fractures as described.25 However, this was not observed in the 129/Sv genetic background, hence OA was induced in the 129/Sv background. It would have been interesting to investigate whether in the DBA/1 the OA phenotype was more severe due to the underlying bone phenotype. However, the lack of load on the knee due to the tibial fracture would have been a confounding element.
OA is a disease of the whole joint, and, in particular, the cartilage and bone form a functional unit that shares cellular and molecular homeostatic mechanisms.31 It is interesting to note how loss of function of the WNT inhibitor FRZB was also associated with increased susceptibility to OA in mice14 and humans,15 but as opposed to WNT16-deficient mice, also with increased bone mass.14 These data stress the importance of the interplay of the cartilage and bone functional unit in OA development.31 Compared with FRZB, WNT16 has some agonist activity and this may explain the opposite phenotype in the bone. It would be interesting to understand the possible redundancies and the contribution of this agonistic activity in the context of joint homeostasis and the respective contribution of these two molecules.
Conditional WNT16 knockout (KO) in cartilage or bone will represent an opportunity to understand the specific roles of WNT16 and of these two tissues in the development of OA.
The fact that, following acute cartilage injury, FRZB is downregulated and WNT16 is upregulated suggests that this difference may play an important role following injury. It would be interesting to understand the possible redundancies and the contribution of this agonistic activity in the context of joint homeostasis and the respective contribution of these two molecules.
Lubricin is essential for joint homeostasis and its disruption results in cartilage breakdown associated with chondrocyte apoptosis, which was also seen in our model and was restricted to the superficial region of the articular cartilage. However, we also noticed that exogenous WNT16 modulated other markers of the SZCs, which produce lubricin. Recent data support the concept that SZCs represent specific progenitor cells in the articular cartilage5 ,6 and that they are mobilised to the site of cartilage damage following injury.32 It is therefore possible that the increased susceptibility to OA of WNT16-deficient mice might be caused not just by the failure to upregulate lubricin following DMM, but by a more general phenotypic derangement of the SZCs, as suggested by the alteration in the expression of multiple markers when stimulated with WNT16. This likely occurs as a result of loss of controlled canonical WNT signalling as in development, the differentiation and maintenance of such important cell population is highly dependent on the maintenance of a balanced activation of canonical WNT signalling at the cartilage surface.3–8
The modulation of canonical WNT signalling by WNT16 is likely the most important element of novelty in this study. Movérare-Skrtic et al25 reported that WNT16 is a weak agonist of the canonical WNT signalling. Here, we confirmed these data and add that it also functions as an inhibitor when administered together with more potent WNT proteins. Both excessive activation8 ,14 ,15 or excessive suppression6 ,7 ,16 ,17 of WNT signalling causes cartilage loss in mice as well as in humans. Therefore, after injury, WNT16 may trigger controlled homeostatic responses, which, in the early phases of OA, have a chance to halt cartilage loss and ensure that the net activation of WNT signalling is sustained so as to allow maintenance of chondrogenic stem cells4–6 but without exceeding the limits that would cause cartilage breakdown. This modality of signalling was not limited to the articular cartilage and OA but was extended to biological contexts as remote as Xenopus development. It is therefore likely that WNT16 may represent a ‘buffering WNT’ in several other contexts when WNT activation is desirable but excessive activation is not, such as in skin wound healing,33–37 and provides an explanation of how a fine balance of WNT activation is achieved in highly dynamic biological contexts such as repair and homeostasis.
Acknowledgments
The authors thank Andrew McMahon (University of Southern California, Los Angeles, California, USA) for providing the wnt16−/− mice, Randall Moon (University of Washington, Seattle, USA) for providing SUPER8XTOPFlash reporter plasmid, Dr Muy-Teck Teh (QMUL, London, UK) for providing a wnt16 expression plasmid and Chris Mole (QMUL, London, UK) for providing BAC. They also thank Frank P Luyten (KU Leuven, Belgium) for critically reviewing the manuscript.
References
Footnotes
Handling editor Tore K Kvien
GN and BLT contributed equally to this manuscript and are co-first authors.
Contributors GN and BLT contributed equally to the manuscript through experimental design, performing experimental work, interpreting results and writing the manuscript. JY, generated the WNT16-deficient mice. OA, SEE and A-ST performed experimental work. JCS contributed to experimental design, performed experiments and interpreted results. LD designed, performed and analysed the Xenopus experiment. GS and JZ designed, performed and analysed the Microct experiment. NE performed experimental work. CP contributed to experimental design and interpretation. FD contributed to experimental design, interpretation of results and writing the manuscript.
Funding The authors thank the Nuffield Foundation (grant QMUL-RHE 36472), the Arthritis Research UK (grants 19654 and 19344), the Medical Research Council UK (MR/K013076/1 and MR/L022893/1), the Medical College of Saint Bartholomews Hospital and the Rosetrees trust (A589) for funding this work.
Competing interests None declared.
Ethics approval All procedures were approved by East London and The City Research Ethics Committee and the UK Home Office.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement This is an open access article distributed in accordance with creative commons attribution non-commercial licence.