Article Text

Statistics from Altmetric.com
A delineation of the differences in pathology between AS and RA
A hallmark of spondylarthropathies (SpA), such as ankylosing spondylitis, (AS) is the fusion of joints as well as intervertebral spaces. This fusion is caused by the formation of bony spurs appearing as syndesmophytes and osteophytes in the intervertebral spaces and in the joints, respectively. Fusion of joints is based on increased endochondral ossification, which allows bone formation and bridges the joint space. Tumour necrosis factor (TNF) is a key proinflammatory cytokine in AS, but is a potent inhibitor of bone formation, and so is unlikely to explain the formation of osteophytes in AS. This is also suggested by recent clinical data showing that TNF blockade seems not to affect structural remodeling of the spinal skeleton in AS, which largely indicates changes due to increased bone apposition. Thus, molecular concepts of structural remodelling in AS need revision, and new pathways involved in bone formation, such as Wingless proteins or transforming growth factor β, might be a clue to the pathogenesis of structural remodelling in AS. The efficacy of TNF blockers to improve clinical symptoms in AS, their poor effect on structural remodelling, and the weak relationship between clinical symptoms and structural damage in AS will profoundly revise our picture of AS in the future.
MECHANISMS OF JOINT FORMATION—MOLECULAR LESSONS FOR JOINT FUSION
Joints and intervertebral spaces form gaps between bones, which allow motion and flexibility. These gaps are actively formed during early development, when chondrogenic formations of the vertebral column and limbs start to branch and build segments. Formation of these gaps depends on the expression of proteins involved in mesenchymal cell differentiation, such as cartilage-derived morphogenic protein 1 (also called GDF5) and bone morphogenic protein (BMP) 5.1 Without these proteins no joints are formed, since the appropriate differentiation of cells, which form the synovial membrane, are then lacking. Wingless (Wnt) proteins, such as Wnt-14 (also known as Wnt-9a), are also crucial for the initiation of joint formation in the limbs.2 Joint formation can thus be considered as an active differentiation process, which replaces the chondrogenic matrix by specific fibroblast-like cells that form the synovial membrane, the periosteum and the joint capsule.
BONY PROTRUSION AS STRESS RESPONSE OF THE JOINT
Joints allow maintaining motion, which, however, requires a structurally intact joint space for smooth gliding of articular surfaces. Inflammation leads to joint damage, which causes pain, swelling, stiffness and functional impairment in patients with chronic inflammatory and degenerative joint disease. Resident mesenchymal tissue in joints, however, is not inert when exposed to an inflammatory attack, and causes certain response patterns, which allow structural remodelling to cope with unphysiological stress. The most prominent pattern is osteophyte formation, which includes spondylophyte and syndesmophyte formation when these structures are located in the axial skeleton. Osteophytes, spondylophytes and syndesmophytes are bony protrusions, which appear on plain radiographs, CT scans and MRI of patients with seronegative SpA, in particular AS, and osteoarthritis (OA), but are virtually absent in rheumatoid arthritis (RA). Syndesmophytes, vertical bony spurs, ultimately leading to a bridge between vertebrae, are a hallmark of AS. Similar lesions, now more horizontally oriented, are also found in degenerative joint diseases such as OA, psoriatic arthritis or haemochromatosis arthropathy, both among vertebral bodies (spondylophytes) and at peripheral joints (osteophytes).
Bony protrusions are based on endochondral ossification, which leads to deposition of the chondrogenic matrix and later to remodelling into bone. Bony spurs emerge from the periosteum close to joints or intervertebral spaces, where mesenchymal cells are localised, which have the ability to differentiate into cartilage and bone, when they receive the appropriate signals. Emergence of osteophytes depends on stress on the joint, and apparently both mechanical stress (as evident from the abundance of such lesions in OA) and inflammatory stress can precipitate their formation. From a pathophysiological point of view these lesions can be seen as an attempt of repair or stabilisation mechanism to reduce motion in the affected joint. Bony spurs can even bridge joints leading to bone ankylosis and complete stabilisation of joints. Longstanding sacroiliitis is a typical example, which, after complete ankylosis and immobilisation of the joint, leads to a marked reduction in clinical symptoms. Bridging syndesmophytes in AS is another clear example.
DIFFERENCES IN INFLAMMATORY BONE REMODELLING BETWEEN RA AND AS
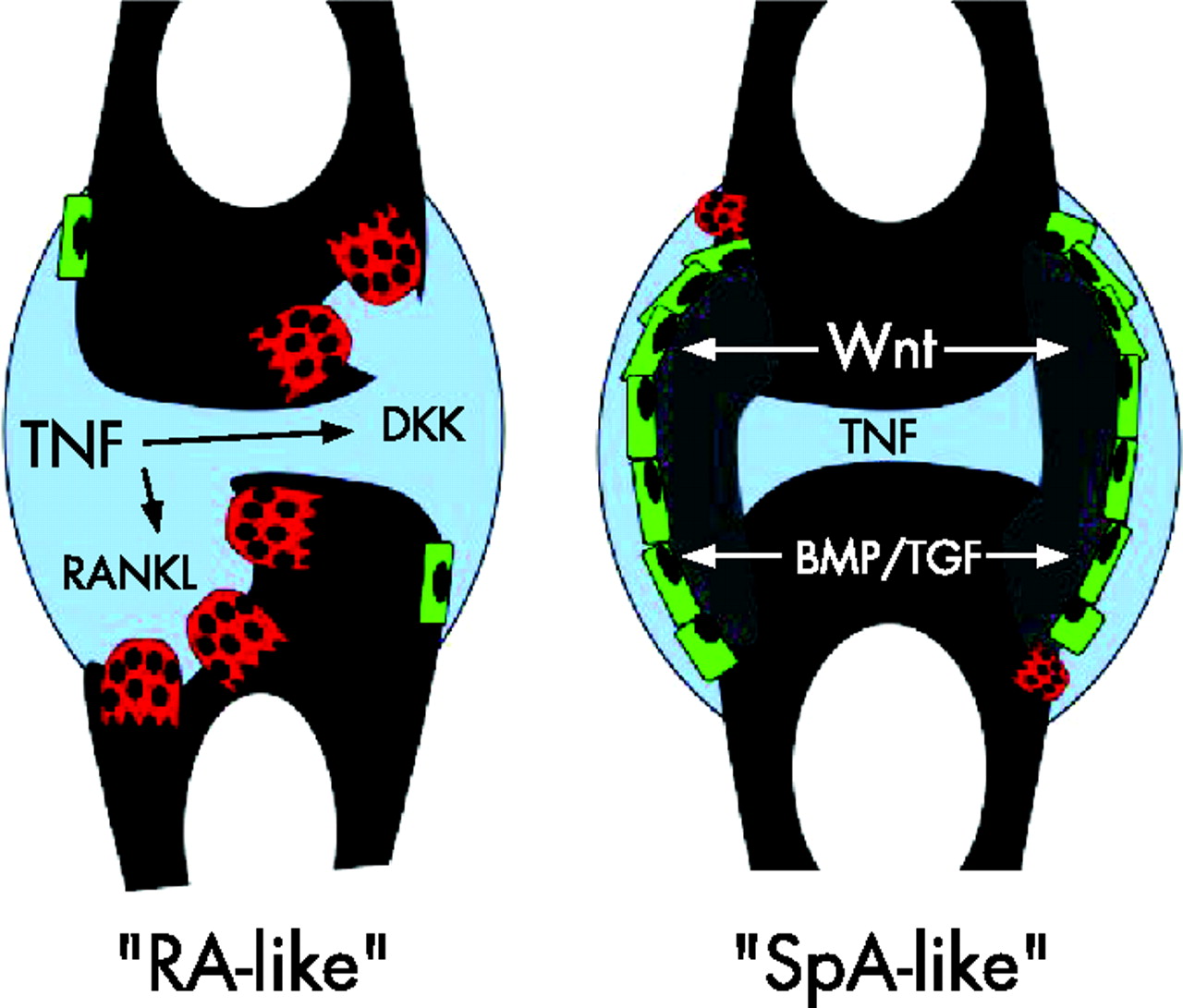
In contrast to AS, RA is the prototype of a disease, which is not associated with osteophyte formation despite severe joint damage (fig 1⇓). The pathophysiological picture of RA is characterised by osteoclast formation and bone destruction, with no or mild signs of bone repair.3,4 This is based on the dominance of bone resorption in RA, which rapidly destroys the periosteal lining and invades the bone. This process is fuelled by rapid generation of osteoclasts through TNF and receptor activator for nuclear factor κ B ligand (RANKL), and enhanced bone resorption combined with a blunted response of bone formation, which involves inhibitors of Wnt proteins, such as Dickkopf-1 (DKK-1).5–,7 The activating role of TNF in osteoclast formation has been defined in the past 5 years, whereas the role of TNF in decreasing osteoblast formation is known for many years but its molecular regulation had been poorly defined until recently.8,9 RA combines rapid bone resorption with inhibition of bone formation leading to unfavourable imbalance of skeletal homeostasis, leading to rapid development of erosions. Structural damage in RA at least partly mimics bone damage of multiple myeloma, since both diseases are characterised by “holes” in the bone with no major reparative response. Importantly, the structural damage measured by radiological scores in RA is a direct consequence of the inflammatory process, which makes the good relationship between clinical disease activity and structural damage in this disease understandable.10

{kind=link}
In rheumatoid arthritis (RA), joints undergo progressive resorption, which is mediated by the generation of osteoclasts (red) in the joint. These cells resorb bone and are induced by receptor activator for nuclear factor κ B ligand (RANKL), which is activated upon tumour necrosis factor (TNF) challenge. Moreover, osteoblasts (green), the bone-forming cells, are suppressed in RA, which is at least partly mediated by Dickkopf (DKK) proteins. In spondylarthopathies (SpA) such as ankylosing spondylitis, bridging of joint and intervertebral spaces by bony spurs (grey) is observed. These changes are based on endochondral ossification and may represent a kind of repair strategy of the joint. TNF drives inflammation in SpA similar to that in RA, but there is no connection to increased bone formation driven by osteoblasts (green), which is based on increased activation of Wingless (Wnt)- proteins, bone morphogenic proteins (BMPs) and transforming growth factor beta (TGFβ). Wnt activation also blocks osteoclast differentiation through increased production of osteoprotegerin.
By contrast, the pathological, and partly also the radiographic, picture of SpA is dominated by response to stress. Despite chronic inflammation, which is a well-known precipitator of trabecular bone loss and also affects patients with AS, there is evidence for a dramatic bone formation in the periosteal compartment in AS but not in RA. It seems that in AS an initial bone-resorptive phase may act as a stress factor, which is followed by profound endochondral bone formation originating from the periosteum and leading to bone spurs that could bridge the joints and fuse the vertebrae. Molecularly these lesions depend on increased bone formation, which is most likely governed by members of the TGF/BMP protein family, as well as by the group of Wnt proteins. Thus, intracellular regulators of TGF-β/BMPs, such as regulatory Smads (Smad6, Smad7), block osteophyte formation in murine joints.11 Added to this, Wnt proteins seem to be regulators of osteophyte formation. Binding of Wnt to its receptor Lrp5/6 and activation of downstream regulators such as β-catenin are crucial for bone formation.12 Moreover, there is a cross-talk with the RANKL–osteoprotegerin system, since Wnt signalling activates osteoprotegerin, which balances RANKL-induced osteoclast activation.13 Inhibitors of Wnt, such as DKK-1, are key target genes of TNF, which probably explains the negative effects of TNF and other proinflammatory cytokines on bone formation. In line with this, DKK-1 levels are low in AS but high in RA, suggesting that the Wnt signalling cascades are turned on in the joints of AS.7 These findings suggest that early developmental programmes are switched on when joints form osteophytes to bridge and stabilise the diseased joint space. It also suggests that TNF negatively regulates bone formation, and that inhibition of bone formation and osteophyte growth by TNF-blocking agents is unlikely.
TNF BLOCKERS AND INFLAMMATORY BONE DESTRUCTION
According to the pathophysiological process explained above, it can be expected that drugs that block the action of TNF are helpful in reducing the formation of new erosions in RA, but that their role in reducing structural damage in AS might be fundamentally different. There is abundant information that the available TNF blockers, etanercept, infliximab and adalimumab, can reduce progression of structural damage and may even lead to repair in RA.14–,16 Moreover, in RA, TNF blockers induce a marked reduction in clinical disease activity, which parallels the reduction in structural damage, but there is also reduction of radiographic progression even in patients with some persistent disease activity, especially if a TNF blocker is used in combination with methotrexate.17 As already indicated above, there is a clear longitudinal relationship between clinical disease activity and subsequent radiographic damage in patients with RA,10,18 as well as a distinct relationship between inflammation on MRI and subsequent structural damage on both MRI and radiographs.19
TNF BLOCKERS AND BONE APPOSITION IN AS
What is known about the relationship between clinical disease activity, findings on MRI and structural damage on radiographs in AS? Very few studies have been published on this topic. From the Outcome in Ankylosing Spondylitis International Study (OASIS) cohort, we know that radiographic progression is unrelated to clinical disease activity parameters and acute-phase reactants; only MMP3 was an independent predictor of radiographic progression in this cohort.20 Other (indirect) data are available from trials using TNF blockers in AS. There is very good efficacy on clinical disease activity, acute-phase reactants and inflammation visible on MRI.21–,23 So far there is only one controlled study (published as abstract) addressing the efficacy of a TNF blocker (etanercept) on progression of structural damage.24 Radiographs from the OASIS cohort were used as a comparator, and there was no difference in radiographic progression over a 2-year time frame between patients from OASIS (without a TNF blocker) and patients treated with etanercept. Possible explanations are differences in disease activity and severity between patients from OASIS and patients included in the trial. However, limiting the analysis to patients from OASIS who would have fulfilled the entry criteria for the trial yielded results that were completely comparable. Also, adjustment for all possible differences in baseline variables did not change the results. Another possibility brought forward was that patients already had established disease, and that reduction in radiographic progression could be seen only during the early phases of the disease. Insufficient duration of follow-up was also considered, but seemed unlikely, as 2 years is already a long duration of follow-up and the clinical efficacy is already seen within 2 weeks. Finally, it was considered whether there might be a difference between etanercept and the two anti-TNF antibodies, infliximab and adalimumab, as there is also a difference in efficacy on inflammatory bowel disease between these agents. However, the above-presented data on pathophysiological processes in bone formation, the major abnormality in AS, give the most likely explanation that it cannot be expected that TNF blockers are effective in reducing new bone formation. Data with longer follow-up, with the antibodies against TNF and in patients with early disease, will give us the final answer whether TNF blockers indeed do not inhibit syndesmophyte formation in AS.
REFERENCES
Footnotes
Competing interests: None declared.