Article Text

Abstract
Objectives To study the efficacy and safety of fasinumab in moderate-to-severe, chronic low back pain (CLBP).
Methods In this phase II/III, double-blind, placebo-controlled study, patients with CLBP aged ≥35 years with inadequate pain relief/intolerance to acetaminophen, non-steroidal anti-inflammatory drugs and opioids were randomised to fasinumab 6 or 9 mg subcutaneous every 4 weeks (Q4W), 9 mg intravenous every 8 weeks (Q8W) or placebo. Primary endpoint was change from baseline to week 16 in average daily low back pain intensity (LBPI) numeric rating score. Key secondary efficacy variables included Roland-Morris Disability Questionnaire (RMDQ) and Patient Global Assessment (PGA). The results are based on a modified intent-to-treat analysis of 563/800 planned patients when enrolment was stopped early given emerging signals of joint risk in other osteoarthritis (OA) studies at doses being tested here.
Results Significant placebo-adjusted LBPI reductions at week 16 were observed for fasinumab 9 mg Q4W and Q8W (least squares mean (standard error) −0.7 (0.3); both nominal p<0.05), but not 6 mg (–0.3 (0.3); p=0.39). RMDQ and PGA improvements to week 16 were greatest for fasinumab 9 mg intravenous. Numerically greater efficacy occurred in patients with, versus those without, peripheral OA (pOA) over 16 weeks. Treatment-emergent adverse events (AEs) occurred in 274/418 (65.6%) patients in the combined fasinumab groups and 94/140 (67.1%) placebo patients. Joint AEs, mostly rapid progressive OA type 1, were more frequent in the combined fasinumab groups (19 events in 16 patients (3.8%) vs 1 event in 1 patient (0.7%) for placebo); all except one occurred in pOA patients.
Conclusions Fasinumab highest doses, but not lower dose, improved both CLBP pain and function. Most joint AEs occurred in pOA patients, consistent with earlier findings in symptomatic OA. Further study is needed of patients with CLBP with and without pOA to determine optimal benefit–risk.
- low back pain
- analgesics
- fibromyalgis/pain syndromes
- osteoarthritis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Inadequate relief of chronic pain has a profound effect on an individual’s quality of life and is associated with substantial healthcare costs and loss of productivity.
What does this study add?
There remains an unmet medical need for alternative treatment options that have analgesic efficacy, mitigate the risks associated with current treatment options and provide an acceptable risk/benefit profile.
Nerve growth factor (NGF) inhibitors have the potential to provide pain relief via a mechanism distinct from that of commonly used analgesic medications such as non-steroidal anti-inflammatory drugs (NSAIDs) and opioids, and thus avoid NSAID or opioid adverse effects such as increased risk of cardiovascular events, gastrointestinal toxicity, drowsiness, respiratory depression, dependence and abuse.
Treatment with NGF inhibitors has been associated with dose-dependent risk of joint damage including rapidly progressive osteoarthritis (OA), that may be more likely in individuals with peripheral OA (pOA) than in those without pOA, and neurologic symptoms, including paraesthesia.
Higher doses were required to relieve chronic low back pain (CLBP) than was observed in previous studies in patients with pain due to hip and knee OA.
How might this impact on clinical practice or future developments?
The results observed in this study support continued evaluation of fasinumab as a possible new treatment option for patients with CLBP with inadequate pain control, or who are intolerant to or have a contraindication for existing therapies.
For future studies in CLBP, consideration will be given to dose of fasinumab to seek the most favourable risk–benefit profile.
Introduction
Low back pain (LBP) is a major international health problem.1 According to the Global Burden of Disease 2017 study, LBP ranked highest among other conditions as measured in disability-adjusted life years.2 Although most patients are believed to recover quickly from acute episodes, recurrence is common.3 Chronic LBP (CLBP) is defined as pain persisting for ≥3 months.4 Guidelines recommend initial treatment with non-pharmacological interventions, including exercise and multidisciplinary rehabilitation.5–8
If these interventions are inadequate or if CLBP persists, guidelines recommend non-steroidal anti-inflammatory drugs (NSAIDs) as first-line pharmacological treatments and duloxetine and tramadol as second-line treatments.7 Stronger opioids are an option only if patients fail the afore-mentioned treatments and if the potential benefits outweigh the risks.7 However, long-term use of both NSAIDs and opioids is limited by tolerability issues and adverse effects, such as gastrointestinal bleeding, cardiovascular events, and the potential for abuse and dependence.9
Neurotrophins are a family of polypeptide growth factors that play a role in the proliferation, differentiation, survival and death of neuronal and non-neuronal cells.10 Nerve growth factor (NGF) was the first neurotrophin identified.11 It provokes pain,12 13 is elevated in the synovial fluid of patients with osteoarthritis (OA)14 15 and its receptors are upregulated in injured and inflamed tissues.16 17 NGF produced by peripheral tissues binds neurotrophic receptors (low-affinity p75 and high-affinity tropomyosin-related kinase A) on nociceptive neurons to modulate pain.18 19 NGF inhibitors might, therefore, provide pain relief via a novel mechanism, potentially avoiding the risks of NSAID or opioids.
Fasinumab is a fully human monoclonal antibody shown to reduce pain in OA.20 21 This study evaluated the efficacy and safety of fasinumab for moderate-to-severe CLBP in patients with intolerance to, or inadequate pain relief from, acetaminophen, oral NSAIDs and opioids.
Methods
Patient and public involvement
This phase II/III, randomised, double-blind, double-dummy, placebo-controlled study (NCT02620020) was conducted at 105 sites in the USA, Canada and Europe.
Study population
Eligible patients were ≥35 years old with CLBP and history of inadequate pain relief or intolerance to analgesic therapy, including acetaminophen, at least one oral NSAID and at least one opioid (or unwillingness to take opioids), and a diagnosis of moderate-to-severe CLBP (Quebec Task Force category 1: no radiating pain, or Quebec Task Force category 2: proximal radiation above the knee)22 for ≥3 months prior to screening. An LBP intensity Numeric Rating Scale (LBPI NRS) score ≥4 at both screening and at randomisation (after withdrawal of previous pain medication(s), without requirement for pain flare), and a Patient Global Assessment (PGA) of LBP of fair, poor or very poor at screening were also required. Presence of OA was not exclusionary (see online supplemental methods for full inclusion and exclusion criteria).
Supplemental material
Study design and treatments
The study consisted of a screening period (up to 30 days), a 7-day prerandomisation period during which all pain medication except study-provided rescue medication was discontinued, a 16-week randomised treatment period and a 20-week follow-up period. Patients were randomised (1:1:1:1) according to a computer-generated central randomisation scheme and assigned by interactive voice response system, to either: fasinumab 6 mg subcutaneously (SC) every 4 weeks (Q4W), fasinumab 9 mg SC Q4W, fasinumab 9 mg intravenously (IV) every 8 weeks (Q8W) or placebo SC Q4W or IV Q8W. Patients randomised to fasinumab 6 mg or 9 mg SC received a loading dose (extra nominal dose) on day 1 (total dose of 12 or 18 mg, respectively), followed by nominal doses at weeks 4, 8 and 12 (total of four doses). Patients randomised to fasinumab 9 mg IV Q8W were not loaded, receiving IV fasinumab 9 mg on day 1 and week 8 (total of two doses). To maintain treatment blinding, patients received double-dummy placebo injections (IV or SC) on days of dose administration.
Randomisation was stratified by baseline LBPI NRS score (<7,≥7), duration of CLBP (<5 years, ≥5 years) and maximum Kellgren-Lawrence (K-L) score (≤2, >2) at any knee or hip joint at screening.
The primary efficacy endpoint was the change from baseline to week 16 in the average daily LBPI NRS score on an 11-point (0–10) NRS. The average daily LBPI NRS score was defined as the average of daily LBPI NRS scores for the 7 days before and including the nominal visit. Secondary endpoints included change from baseline to weeks 2, 4, 8, 12 and 16 in Roland-Morris Disability Questionnaire (RMDQ) total score and PGA score, and change from baseline to weeks 2, 4, 8 and 12 in LBPI NRS score.
In October 2016, the US Food and Drug Administration (FDA) placed the study on partial clinical hold following a single case of rapidly progressive osteoarthritis (RPOA) that occurred in a patient with knee OA (K-L score 3 at screening), prompting review of study entry criteria. Since patients with concomitant OA could have received fasinumab doses that had been eliminated by the sponsor from an ongoing fasinumab phase III OA study (NCT02683239) due to the rate of arthropathy, the FDA required that the protocol be amended to either exclude patients with peripheral OA (pOA) or to lower the doses to be studied. Since 70% of the target sample (563/800 patients) had already been randomised, investigators and relevant health authorities were notified that the sponsor stopped enrolment and any further dosing. The statistical analysis plan was updated prior to database lock and a final analysis was performed on completion of all protocol-described study visits, to allow assessment of safety and efficacy, including subgroup analyses of the primary and secondary endpoints by pOA status.
Safety assessments
The safety and tolerability of fasinumab compared with placebo was assessed by treatment-emergent adverse events (TEAEs) during treatment (including the day from first dose of study drug to 4 weeks after last dose of SC study drug or 8 weeks after last dose of IV study drug, whichever was later) and post-treatment (up to 20 weeks) adverse events (AEs). Joint and general safety were monitored independently (see online supplemental methods) as previously described.21
Statistical analysis
The primary efficacy endpoint, change from baseline to week 16 in average daily LBPI NRS score, was analysed using a mixed-effect model repeated measures approach based on the modified intent-to-treat (mITT) analysis set, according to a prespecified analysis established prior to database lock, in response to the early termination of dosing in the study. The mITT analysis set included all randomised patients who received at least one dose of allocated treatment, including all data available up to 5 weeks (4 weeks visit interval + 1 week allowable visit window) after the last dose of study drug. Analyses were deemed exploratory (all p values are nominal). Further details are provided in the online supplemental methods.
The safety analysis set included all randomised patients who received any study drug. Sensitivity analyses for the primary and secondary endpoints used the full analysis set (all randomised patients). Assuming a significance level of 0.05 and a 20% dropout rate by week 16, an enrolment of 200 patients per treatment group would provide at least 91% power to detect a treatment difference of 0.9 between fasinumab 9 mg SC Q4W and placebo for the primary efficacy endpoint with a common SD of 2.4.
To assess potential differences in efficacy and safety between those with and without pOA at baseline, subgroup analyses were performed on the primary and secondary efficacy endpoints, using medical history and/or radiographic evidence of OA (K-L score ≥2 at the hip or K-L score ≥3 at the knee based on screening radiographs), in line with key components of the American College of Rheumatology OA criteria.23 Subgroup analyses for the primary efficacy endpoint were also conducted for randomisation strata (baseline LBPI NRS score (<7, ≥7), duration of chronic LBP (≥5 years, <5 years) and maximum K-L score in any knee or hip joint (≤2,>2)).
Results
Overall, 1783 patients were screened; 563 patients were randomised (figure 1). Patient demographic and baseline characteristics were generally balanced across groups (table 1). Most patients (82.2%) had a maximum K-L score at any knee or hip joint of ≤2 at screening; 14.7% and 3.0% of patients had scores of 3 and 4, respectively, (table 1). Of 558 patients who received at least one dose of study drug (safety analysis set), 35.3% to 42.4% of the fasinumab SC groups and 36.4% of the placebo SC group received all planned doses through week 16; corresponding values for IV groups were 54.3% (fasinumab 9 mg Q8W) and 51.4% (placebo) (online supplemental table 1).

Patient disposition. EOT, end of treatment; FDA, US Food and Drug Administration; IV, intravenous; mITT, modified intent-to-treat; Q4W, every 4 weeks; Q8W, every 8 weeks; SC, subcutaneous; W, week. *Includes, among other reasons: out of screening window, study stopped by sponsor; patients could be excluded for >1 reason.
Demography and baseline characteristics (full analysis set)
Efficacy
Baseline LBPI scores were comparable across treatment groups (table 2). Significant reductions versus placebo in LBPI scores from baseline to week 16 were observed in the fasinumab 9 mg SC Q4W and 9 mg IV Q8W groups (least squares mean (standard error) −0.7 (0.3), nominal p=0.02; and −0.7 (0.3), nominal p=0.03, respectively), but not for the 6 mg SC Q4W group (–0.3 (0.3); nominal p=0.39) (table 2). Pain scores improved as early as week 2 (figure 2A). At week 8, all fasinumab doses provided reductions in LBPI scores versus placebo (least squares mean (standard error) 6 mg SC –0.5 (0.3), nominal p=0.04; 9 mg SC Q4W –1.1 (0.3), nominal p<0.01; 9 mg IV Q8W –1.0 (0.3), nominal p<0.01) (table 2). Mean baseline RMDQ (10.7 to 11.7) and PGA (3.4 to 3.5) scores were comparable across groups. RMDQ reductions were observed as early as week 2 in all fasinumab groups versus placebo and maintained to week 16, with the greatest reductions in the 9 mg IV group (table 2 and figure 2B,C). Placebo-adjusted changes in RMDQ at week 16 were –2.2 to –2.5 across fasinumab groups (all nominal p<0.01). Placebo-adjusted changes in PGA at week 16 (–0.1 to –0.3) reached significance only for fasinumab 9 mg IV (nominal p=0.01).
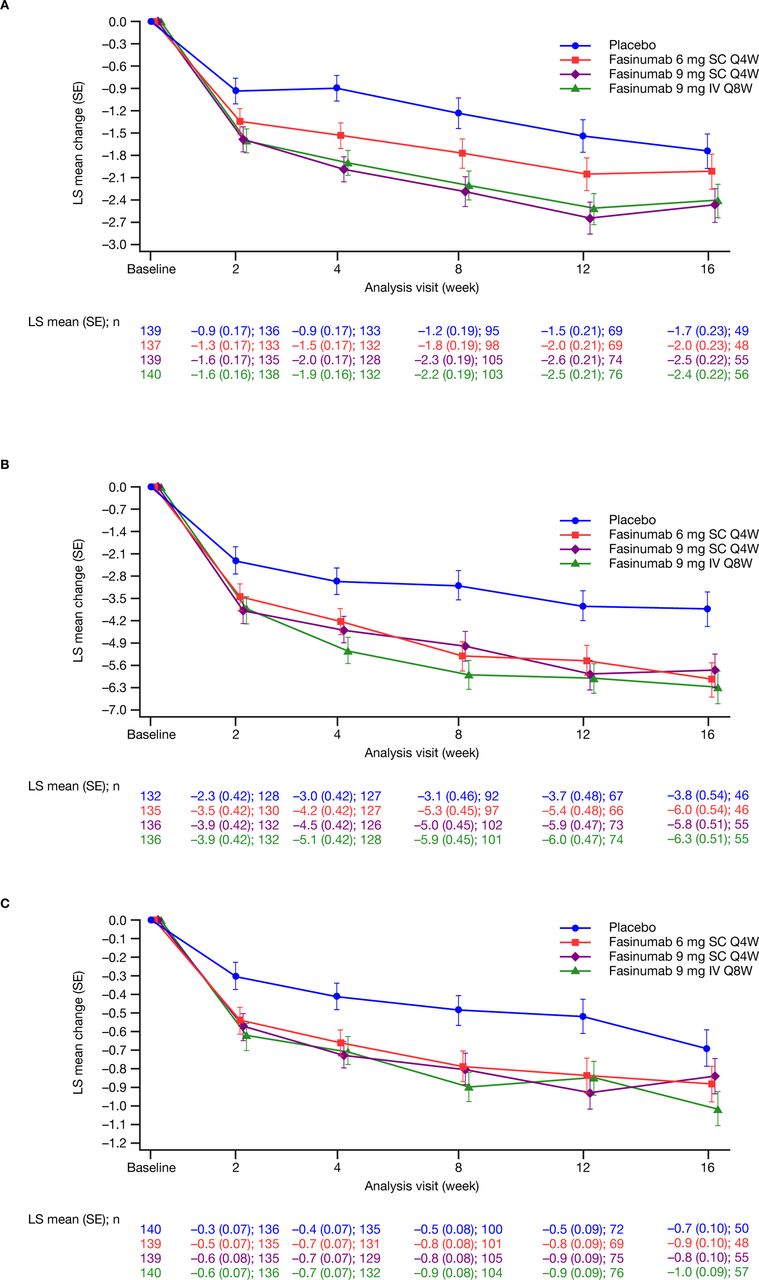
Least squares mean change from baseline in (A) average daily Lower Back Pain Intensity Numeric Rating Scale score (B) Roland-Morris Disability Questionnaire total score (C) Patient Global Assessment of lower back pain score by study visit (modified intent-to-treat analysis set). Analyses are based on mixed effect model repeated measures with baseline randomisation strata, baseline, treatment, visit and treatment-by-visit interaction. IV, intravenous; LS, least squares; Q4W, every 4 weeks; Q8W, every 8 weeks; SC, subcutaneous.
Change from baseline to week 8 and week 16 in the average daily LBPI NRS, RMDQ and PGA of LBP scores (mITT analysis set)
Subgroup analyses
In patients with (57.2%) and without (42.8%) pOA, placebo-adjusted improvements in LBPI scores were greatest in the 9 mg dose groups from week 2 through week 16 (online supplemental table 2 and online supplemental figure 1). Improvement versus placebo was generally numerically greatest in patients with, versus those without, pOA over the 16 week treatment period, with greater separation seen between the pOA subgroups at earlier time points when more patient data were available. A similar pattern was observed for RMDQ and PGA scores (online supplemental table 2 and online supplemental figures 2 and 3). Placebo-adjusted LBPI scores from baseline to week 16 were consistent across randomisation strata (data not shown).
Safety
On treatment, the percentages of patients with ≥1 TEAE were similar between placebo (67.1%; n=94) and combined fasinumab groups (65.6%; n=274), and across the fasinumab dose groups (online supplemental table 3). The system organ class (SOC) with the highest incidence of TEAEs while on treatment was musculoskeletal and connective tissue disorders (16.0% for combined fasinumab groups and 22.1% for placebo) (table 3). Arthralgia was the only TEAE reported in >10% of patients in any treatment group, with a similar incidence in the placebo and combined fasinumab groups (12.1% and 12.4%, respectively). TEAEs of paraesthesia were more frequent for the combined fasinumab groups than for placebo (5.7% vs 2.9%); most of these events were of mild-to-moderate severity. During the post-treatment follow-up period, the overall incidence of AEs in the combined fasinumab groups (29.9%) was similar to that for placebo (27.9%) (online supplemental table 4).
TEAEs with >3% incidence by system organ class and preferred term during the on-treatment period (safety analysis set)
In total, 16 serious AEs (SAEs) occurred in 14 patients (placebo, n=4 (2.9%); combined fasinumab groups (n=10 (2.4%)) during the on-treatment period (online supplemental tables 3 and 5). Three SAEs were considered related to study drug, two of which were in the fasinumab 9 mg groups (haemorrhagic stroke and meniscus injury). In total, 35 SAEs occurred in 31 patients in the post-treatment follow-up period (online supplemental table 6); most were in the SOC of musculoskeletal and connective tissue disorders, all of which occurred in the fasinumab groups (5 patients, 3.6%, in the 9 mg IV group, and 3 patients, 2.2%, in each SC group; 11 total patients, 2.6%). Within this SOC, the most frequent SAE was RPOA. One patient (fasinumab 6 mg) with a history of smoking died of small cell lung cancer during the post-treatment follow-up period (considered unrelated to study drug).
AEs of special interest included sympathetic nervous system dysfunction and adjudicated arthropathies (AAs). No confirmed cases of the former were observed. There were 20 joints with AAs in 17 patients (table 4). All except one AA were detected outside of the prespecified on-treatment period (online supplemental figure 5), and all but one occurred in the fasinumab groups. Of the 20 joints with AAs, 19 were in patients in the pOA subgroup (table 4), and most (15/20) occurred in joints with screening K-L scores of ≥2 at the knee or hip (online supplemental table 7); in 3 knee joints, the screening K-L score was 0 or 1; in 2 shoulder joints, K-L score was not assessed but screening radiographs documented 1 with moderate OA and 1 with severe OA). Adjudicators could report more than one AA category per joint. Most AAs were categorised as RPOA (ie, RPOA type 1 or 2) and among these, 14 joints had RPOA1 (X-ray joint space narrowing; cartilage loss by MRI) solely; 2 joints in two patients (6 mg SC and 9 mg IV) had an RPOA2 (bone fragmentation or destruction; one with RPOA1); 1 joint had subchondral insufficiency fracture (SIF) as the sole finding (9 mg IV); and three joints had SIFs in conjunction with RPOA1. Only two AAs (one RPOA1; (9 mg SC), and one RPOA2; (9 mg IV)) were detected on imaging prompted by symptoms; others were detected on scheduled images. No primary osteonecrosis was observed.
Summary of AAs across the treatment period and post-treatment follow-up period (safety analysis set)
Four joint replacements (knee) were performed in four patients. Two of these occurred following detection of an AA (9 mg SC, RPOA1; 9 mg IV, RPOA2). For the remaining two, preoperative imaging did not detect AA. In one case (9 mg SC), joint replacement was pursued to address functional consequences of pre-existing OA; in the other case (placebo), it was based on need for revision surgery related to historical hemiarthroplasty.
An increase in mean alkaline phosphatase (ALP) occurred over time in all three fasinumab groups (figure 3). The extent of the increase was similar across groups and small compared with baseline values. A small number of patients had increases in ALP above the upper limit of normal (ULN; 150 U/L): placebo (n=3), fasinumab 6 mg (n=2), 9 mg SC (n=4) and 9 mg IV (n=3), none of which met the prespecified definition for potential clinical significance (≥1.5×ULN). During the 20-week post-treatment follow-up period, mean ALP values returned towards baseline (figure 3). A similar pattern was observed for patients with and without pOA (online supplemental figure 4).

{kind=link}
{kind=link}
{kind=link}
Mean change from baseline in alkaline phosphatase (U/L) (safety analysis set). IV, intravenous; Q4W, every 4 weeks; Q8W, every 8 weeks; SC, subcutaneous.
Treatment-emergent antidrug antibody (ADA) responses occurred in five patients (1.3%) on fasinumab and one patient (0.8%) on placebo. All ADA-positive patients exhibited low titre responses, and none was neutralising. A positive ADA response did not affect concentrations of fasinumab.
Discussion
In this study, fasinumab provided improvements in CLBP, function and overall patient assessment of benefit. Although these outcome measures were focused on assessment at the end of the 16-week treatment period (primary endpoint), improvements were noted across most parameters and dose regimens as early as week 2. A key limitation of the study is that because of the FDA hold and early termination of dosing, results are based on an incomplete cohort (35%–56% receiving all planned doses of study drug); data for fewer patients than originally planned were available for efficacy and safety analyses, and p values were considered nominal. Exposure data are limited because of the relatively short treatment duration (16 weeks) and because not all subjects received all planned doses. Moreover, the pOA subgroup analyses were exploratory (a formal diagnosis of OA per American College of Rheumatology (ACR) criteria was not required at study entry and patients were not stratified for OA), but provided an opportunity to address emerging concerns about AA risk in pOA patients as the development programme matured. The use of loading doses in the fasinumab SC groups should also be considered when interpreting the efficacy and safety results. Although the loading dose may have influenced SC treatment effect at earlier time points, it did not seem to impact differences in effect noted across the 9 mg SC and IV dose groups, though this possibility cannot be excluded for the 6 mg SC group.
Although cross-study comparisons are imprecise, the placebo-adjusted treatment effect of fasinumab at endpoint, which ranged from –0.3 for fasinumab 6 mg SC Q4W to –0.7 for 9 mg SC Q4W and IV Q8W, is broadly consistent with studies in patients with CLBP of another NGF inhibitor, IV or SC tanezumab, which reported week-16 placebo-adjusted treatment effects of –0.3 for 5 mg and –0.4 to –0.8 for 10 mg.24 25 The efficacy of fasinumab in the current study also appears comparable or slightly better than most potent opioids (placebo-adjusted treatment effect of –0.4 was reported in a systematic review and meta-analysis),26 and maximal doses of NSAIDs (treatment effect of –0.4 for naproxen was reported in the IV tanezumab trial).25
CLBP can be caused by various aetiologies including chronic muscular pain, discogenic pain and facet joint OA. However, prior studies have been unable to deconvolute the various components that might contribute to pain in different patients. Our study provided an opportunity to evaluate responses across subgroups without and with pOA, known to be associated with facet joint OA.27 Since studies focused on OA had suggested a dose-related risk of arthropathy,20 analysis by pOA status was also an opportunity to uncover differential safety patterns in the treatment of CLBP. Patients with pOA generally experienced greater placebo-adjusted improvement in pain and function than those without, in part driven by greater resolution of pain in the placebo group of the non-pOA subgroup, and was particularly evident at earlier timepoints (4 and 8 weeks), when more patient data were available for assessment. These findings might reflect differential components of pain in these two subgroups. For example, a greater proportion of patients without pOA may have had CLBP caused by factors other than OA of the spine, such as proximal radiculopathy (ie, included in Quebec Task Force category 2). NGF inhibitor therapy has shown no benefit in patients with pain caused by radiculopathy (ie, sciatica).28
The incidence of TEAEs was similar between placebo and fasinumab. However, patients treated with fasinumab had higher rates of AAs across all doses studied. All but one AA occurred in patients with concomitant pOA, suggesting that pOA patients may be more predisposed than those without to risk of arthropathy at the high fasinumab doses used in this CLBP study. These findings are consistent with studies that reported higher rates of arthropathy at the highest doses of fasinumab and tanezumab, beyond those producing maximal treatment benefit in OA pain.20 25 29–31 Across a higher dose range studied in CLBP here, there was no clear difference across doses in the frequency of AA events, even when focusing only on the pOA subgroup.
Elevations in ALP with fasinumab treatment were observed in a phase IIb/III study in patients with OA of the knee or hip.20 In the current study, mean ALP elevations (peak at week 16) were lower than observed in the previous OA study, even in the pOA subgroup. ALP levels returned towards normal during the post-treatment follow-up, as has been previously reported.20 It is unclear whether these small changes in ALP associated with treatment represent bone turnover or a more independent effect on enzyme production or enzymatic activity.
Despite dosing being prematurely terminated, all fasinumab doses provided improvements versus placebo in measures of pain (average daily LBPI NRS score), function (RMDQ) and overall patient assessment (PGA) over the first 8 weeks of the study. Significant pain improvement was maintained over 16 weeks for both fasinumab 9 mg groups, but not for 6 mg. Further studies will be needed to determine whether the robust efficacy shown at week 8 is sustained for longer durations at lower doses. Although the treatment benefit in this study was numerically greater in the pOA subgroup, the rates of AA in these patients were substantially higher. This study, therefore, validated concerns about the use of fasinumab in CLBP subjects with concomitant OA, whose benefit–risk at the highest doses was unacceptable. For patients without pOA, low rates of AA were observed at these high doses, though treatment effect was more modest. In these patients, since their back pain may be dominated by mechanisms other than OA, fasinumab may be less likely to provide benefit. Hypothetically these patients may also need even higher doses, and joint AEs would need to be carefully balanced against treatment benefits. Further studies with longer treatment and follow-up are needed to inform benefit–risk.
Acknowledgments
The authors and sponsor would like to thank the patients, their families and all investigators involved in this study. Medical writing assistance and editorial support, under the direction of the authors, was provided by Michele Damo, PharmD, of Prime (Knutsford, UK), funded by Regeneron Pharmaceuticals, and Teva Pharmaceutical Industries according to Good Publication Practice guidelines (http://annals.org/aim/article/2424869/good-publication-practice-communicating-company-sponsored-medical-research-gpp3). The sponsors were involved in the study design, and collection, analysis and interpretation of data, as well as data checking of information provided in the manuscript. The authors had unrestricted access to study data, were responsible for all content and editorial decisions, and received no honoraria related to the development of this publication.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Josef S Smolen
Contributors PD, AJK, JG, NS, SJD, CAE, HG, NS, DMW, GDY and GPG contributed to the study design or concept and the interpretation of the data, and critically reviewed and edited the manuscript. CAE and HG performed the statistical analyses. All authors approved the final version.
Funding This study was funded by Regeneron Pharmaceuticals and Teva Pharmaceutical Industries.
Competing interests PD, SJD, CAE, HG, GPG, NS, DMW and GDY are employees of Regeneron Pharmaceuticals. JG reports consulting fees from Pfizer and participation in other activities with Regeneron Pharmaceuticals outside the submitted work. AJK reports participation in other activities with Altoona Center for Clinical Research, PC, during the conduct of the study; and other activities from AbbVie, Celgene, Horizon, Janssen, Merck, Novartis, Pfizer, UCB, Genzyme, Sanofi, Regeneron Pharmaceuticals, SUN Pharma Advanced Research, Boehringer Ingelheim, Flexion, Amgen and Gilead, outside the submitted work. NS reports grants from Regeneron Pharmaceuticals during the conduct of this study; and personal fees from Regeneron Pharmaceuticals and Orthofix, outside the submitted work.
Patient consent for publication Not required.
Ethics approval The protocol was approved by local institutional review boards and/or ethics committees (see online supplemental methods) and conducted in accordance with the ethical principles outlined in the Declaration of Helsinki, consistent with Good Clinical Practices and applicable regulatory requirements. Informed consent was obtained from each patient.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Qualified researchers may request access to study documents (including the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan) that support the methods and findings reported in this manuscript. Individual anonymized participant data will be considered for sharing once the product and indication has been approved by major health authorities (e.g., FDA, EMA, PMDA, etc), if there is legal authority to share the data and there is not a reasonable likelihood of participant re-identification. Submit requests to https://vivli.org/
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.