Article Text

Abstract
Technology for precise and efficient genetic editing is constantly evolving and is now capable of human clinical applications. Autoimmune and inflammatory diseases are chronic, disabling, sometimes life-threatening, conditions that feature heritable components. Both primary genetic lesions and the inflammatory pathobiology underlying these diseases represent fertile soil for new therapies based on the capabilities of gene editing. The ability to orchestrate precise targeted modifications to the genome will likely enable cell-based therapies for inflammatory diseases such as monogenic autoinflammatory disease, acquired autoimmune disease and for regenerative medicine in the setting of an inflammatory environment. Here, we discuss recent advances in genome editing and their evolving applications in immunoinflammatory diseases. Strengths and limitations of older genetic modification tools are compared with CRISPR/Cas9, base editing, RNA editing, targeted activators and repressors of transcription and targeted epigenetic modifiers. Commonly employed delivery vehicles to target cells or tissues of interest with genetic modification machinery, including viral, non-viral and cellular vectors, are described. Finally, applications in animal and human models of inflammatory diseases are discussed. Use of chimeric autoantigen receptor T cells, correction of monogenic diseases with genetically edited haematopoietic stem and progenitor cells, engineering of induced pluripotent stem cells and ex vivo expansion and modification of regulatory T cells for a range of chronic inflammatory diseases are reviewed.
- inflammation
- autoimmunity
- autoimmune diseases
Statistics from Altmetric.com
Introduction
Many chronic human autoimmune and autoinflammatory diseases require indefinite therapy. Despite major developments in molecular mechanism-focused therapy for rheumatic conditions in the past several decades, substantial morbidity and mortality still attends these diseases. Emerging gene therapy approaches offer the promise of more specific and durable treatments that may circumvent toxicities of traditional medical management for inflammatory disease. Rare periodic fever syndromes with monogenic causes are clear candidates for potential application of genetic modification therapy. Of course, the aetiology of most chronic inflammatory disorders is complex, involving contributions of multiple genetic and environmental factors. Nonetheless, the explosive growth in technology for precise and robust genetic editing over the past decade has generated interest in using targeted genetic modifications for the treatment of these acquired polygenic inflammatory diseases as well.
The principle of gene editing involves deliberate and specific modifications to genomic DNA or mRNA with the intent to modify gene product expression, structure or function. Chromosomal DNA modifications rely primarily on the ability to generate double-stranded breaks (DSB) that serve to localise targeted changes to the effected DNA. Clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated protein (CRISPR/Cas9) has rapidly become the tool of choice to this end, but older techniques such as zinc-finger nucleases (ZFN) and transcription activator-like effector nucleases (TALEN) have been used extensively. New applications of the nuclease Cas9 have the potential to greatly expand our ability to manipulate nucleic acids and protein expression, structure and function. Emerging technologies such as base editing, activation or repression of transcription via Cas9-targeted transcription factors or Cas9-targeted epigenetic modifiers and RNA editing are novel technologies that could greatly expand our ability to discretely control pathobiology. Viral and non-viral vectors can be employed to deliver effectors of genome editing to the cell or tissue of interest, including T cells, haematopoietic stem cells (HSC), induced pluripotent stem cells (iPSC) and organ parenchymal cells.
Genome editing
The overall goal of genome editing is to alter effector protein expression, structure or function. Gene editing has traditionally been used to this end and facilitates efficient and accurate modification of DNA at a specific locus or loci by generation of a DSB.1 DSBs are then modified by endogenous cellular machinery that mediates either homologous recombination (also termed ‘homology-directed repair [HDR]’), or non-homologous end joining (NHEJ). HDR repairs DSBs by transfer of the modified or wild-type DNA into its homologous target location in the genome. Despite prior extensive use, targeted modification of genomic DNA by HDR has been limited by relative inefficiency, high rates of random template DNA insertion, off-target insertion, and function only during S and G2 cell cycle phases.2–4
NHEJ, in contrast, is the most common manner of DSB repair in mammalian cells, is active during all phases of the cell cycle5 and does not require a homologous template for repair. Due to the latter, NHEJ can create insertions and deletions of varying sizes (termed ‘indels’) at the repair site, which can mutate the protein coding sequence in a negative fashion. Thus, the method of DSB repair is an important consideration when crafting experimental techniques for genome editing.
Investigation of inflammatory disease pathobiology has flourished with the modern capabilities of genome editing. Using conventional and conditional targeted murine alleles, as well as reporter genes, and mice humanised to express human genes and gene variants, the biology of major inflammatory states, such as rheumatoid arthritis and systemic lupus erythematosus, has been rigorously addressed both systemically and at the tissue level. In some instances, this has led to the development of highly potent targeted therapies. Modern techniques for manipulation of the genome are increasingly precise, efficient and facile. The rapid development and capability of the technology will undoubtedly foster increasingly insightful biologic discovery and efficacious therapies for both immune and inflammatory disorders. In this section, we provide a brief history of older tools of genome editing, many of which are still used today, and review state-of-the-art technologies for genetic manipulation of human inflammatory disorders (table 1).
Technologies for genetic editing and their relative advantages and disadvantages based on the existing literature
Older tools for genetic editing
Single-stranded oligonucleotides and meganucleases were initial forays into gene editing. Both are inefficient, difficult to use and have variable specificity.6–11 ZFNs are artificially engineered endonucleases that represented a significant step forward in efficiency and capability.12 13 They have been used in multiple applications in human and animal models.14–16 However, there is lingering concern over ZFN off-target effects,17 and the time, difficulty and sophisticated laboratory methods required for their design and construction.
TALENs are an evolution of ZFNs18 and are notable for their high rates of DNA cleavage and limitless target range.19 They too have been used in a wide variety of organisms.19 However, TALENs share many drawbacks with other endonucleases, including the capacity for off-target effects,19 inefficient delivery to target cells due to large size and epigenetic modifications on the target chromosome interfering with successful HDR.20
CRISPR/Cas9
The CRISPR/Cas9 system is a defence mechanism found in bacteria and archaea.21 22 CRISPR sequences consist of small unique ‘spacer’ sequences, which are interspersed in stretches of highly conserved repetitive DNA sequences, termed ‘CRISPR repeats’, typically located next to groups of highly conserved protein-coding genes called CRISPR-associated (cas) genes that often carry domains similar to nucleases, helicases, polymerases and nucleotide-binding proteins.21 22 The combination of spacer sequences complementary to intracellular but foreign nucleic acids and the Cas9 nuclease fosters degradation of the foreign genetic material and protection from pathogens.
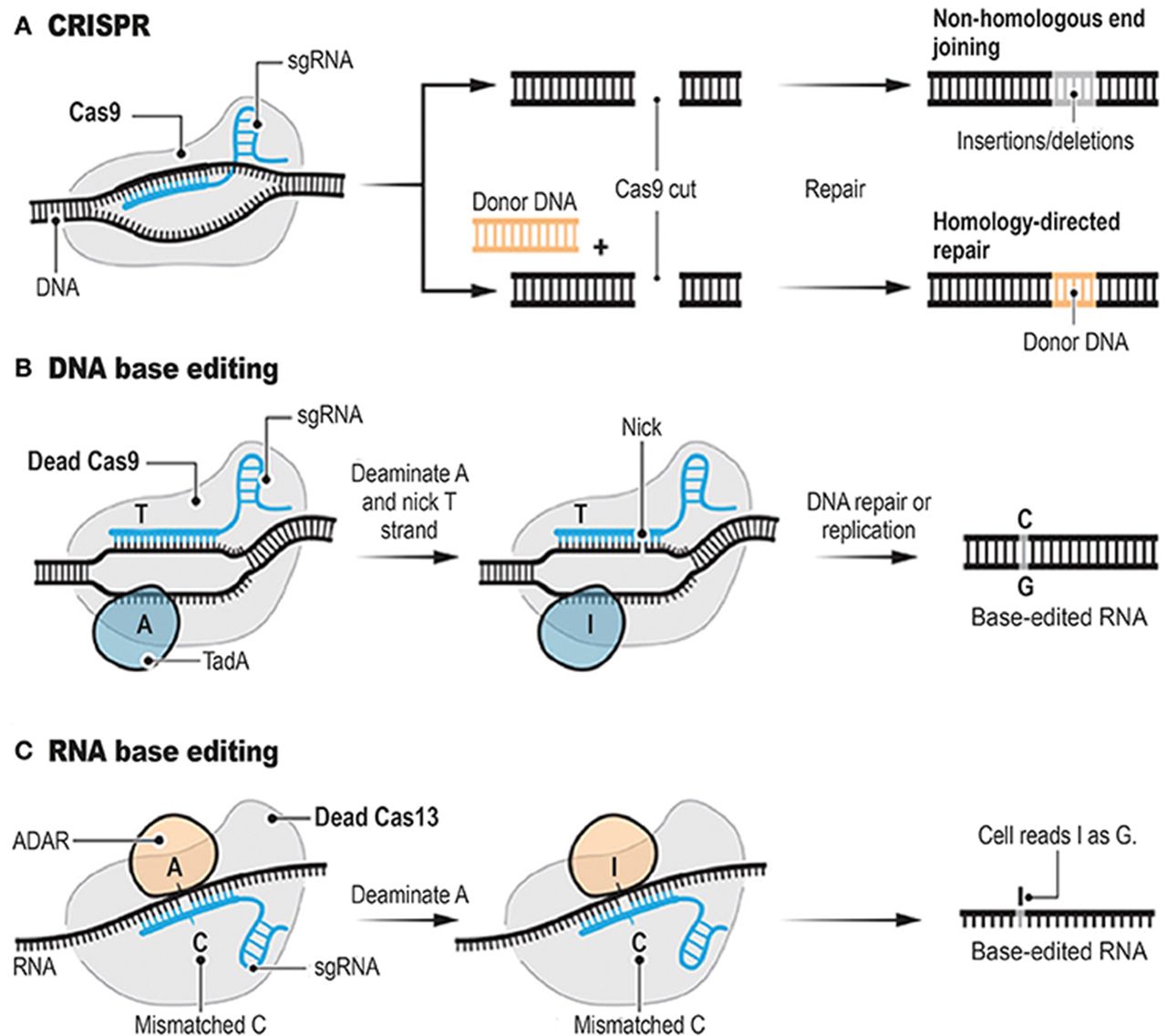
The CRISPR/Cas9 system employs two components for creating DSBs—a customisable single-stranded guide RNA (sgRNA) and an endonuclease (figure 1). The sgRNA is made up of a precursor CRISPR RNA (crRNA) that contains the full length of CRISPR repeats and embedded spacer sequences23 that hybridise to a separately transcribed complementary trans-activating crRNA. The sgRNA guides the Cas9 enzyme, a double-stranded RNA-specific ribonuclease, to any target site.24–27 The RNA-guided Cas9 enzyme surveys the genome, recognising conserved three-nucleotide species-specific protoadjacent motifs (PAM) in the target genome. On binding, the Cas9-gRNA complex detects the DNA complementarity with the guide RNA and creates site-specific DSBs to generate a blunt end usually at a position 3 bp before the PAM sequence. This DSB is then made amenable to repair by either NHEJ or HDR.28–30

The clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 system for targeted genome editing. Initially, RNA-guided Cas9 generates a blunt-ended DSB 3 bp upstream of the PAM sequence. DSBs are then repaired either by NHEJ-mediated disruption or by HDR-mediated modification of the genome. DSBs induced by Cas9 (yellow) can be repaired in one of two ways. In the error-prone NHEJ pathway, the ends of a DSB are processed by endogenous DNA repair machinery and rejoined, which can result in random indel mutations at the site of junction. Alternatively, a repair template in the form of a plasmid or ssODN can be supplied to leverage the HDR pathway, which allows high fidelity and precise editing. Single-stranded nicks to the DNA can also induce HDR. DSB, double-stranded break; HDR, homology-directed repair; NHEJ, non-homologous end joining; PAM, protoadjacent motif; sgRNA, single-stranded guide RNA; ssODN, single-stranded oligodeoxynucleotide. (Reproduced from Ran et al 32 with permission; © Nature Publishing Group)
CRISPR/Cas9 is versatile, efficient, simple to design and use, increasingly specific and is rapidly supplanting other modalities of gene editing.26 Moreover, its functionality is also being broadened. Using multiple gRNAs with the same Cas9 nuclease allows for targeting multiple genes simultaneously.30 31 A single amino acid substitution in either of the two-nuclease domains of Cas9 results in a ‘nickase’ that cleaves only one strand of DNA,32 potentially reducing off-target effects. The refinements of CRISPR/Cas9 and other microbial endonucleases33 34 have resulted in CRISPR/Cas9 rapidly becoming the workhorse for a range of applications including gene therapy, functional genomic screening, transcriptional modulation and synthetic biology.
Base editing with Cas9
Historically, genetic modification has relied on the ability to create a DSB at a specific site of interest and to use the endogenous cell repair machinery for HDR or NHEJ (figure 2A). Methods that induce DSBs have been plagued by ‘off-target’ random insertions or deletions (indels) of variable predictability, and by low efficiency under circumstances practical for therapeutic development, including modification of non-dividing cells.30 32 Komor et al designed and tested a novel method of generating single base pair changes without first generating a DSB.35 The group recognised the capacity of ‘dead’, enzymatically inactive, Cas9 to retain its ability to target and bind DNA in a guide RNA-directed manner. They sought to couple this function with the ability to make a base substitution without a DSB (figure 2B). They then used cytidine deaminases to catalyse the deamination of cytosine (C) to uracil (U), which binds complementary bases like thymine (T). This method is synergistic with the effects of Cas9 in that on binding of the sgRNA/Cas9, nine nucleotides of DNA are unpaired to a single strand,36 with single strandedness being a requirement for cytidine deaminase activity.37 Using a chimeric fusion protein comprising rat-derived cytidine deaminase APOBEC1 fused with nuclease-deficient Cas9, the group observed base editing efficiency rates as high as 37% with a 1.1% indel formation rate. These rates compared favourably with standard wild-type Cas9 and sgRNA, reagents which averaged 0.5% efficiency with a 4.3% indel formation rate. A second form of fusion protein was slightly less efficient but with even lower indel formation rate. Proof of principle experiments showed that human disease-associated mutations could be corrected in mouse astrocytes and human breast cancer cell lines using base editing techniques.

Programmable genome editing tools. The conventional CRISPR DNA editor functions via the double-stranded break (DSB) (A). CRISPR relies on the ability of CRISPR sgRNAs to target the Cas9 endonuclease to precise genomic locations, where Cas9 introduces DSBs. Base editors borrow sgRNAs and Cas9 or other nucleases from CRISPR. However, base editors do not cut the double strand, but instead they chemically alter single bases with deaminase enzymes such as TadA (B, DNA base editor) and ADAR (C, RNA base editor). ADAR, adenosine deaminase, RNA specific; CRISPR, clustered regularly interspaced short palindromic repeats; sgRNA, single-stranded guide RNA; TadA, tRNA-specific adenosine deaminase. (Reproduced from Cohen122 with permission from The American Association for the Advancement of Science)
RNA editing
There is considerable interest in using the specificity of Cas9 and related nucleases in conjunction with sgRNAs for transient, targeted modification of gene expression, structure or function. Post-transcriptional modification of mRNA is natural biologic process and synthetic modifications would be limited by the degradation of the mRNA and subsequently translated proteins. Cox et al described a method harnessing the specificity of CRISPR-associated RNA-guided ribonuclease Cas13 fused to adenosine deaminase acting on RNA 2 (ADAR2) for targeted modification of specific RNAs by deamination of adenosine to inosine, which is equivalent to guanosine in translation and splicing.38 The investigators created a mutated ADAR2 deaminase domain with relaxed sequence constraints to increase editing efficiency and fused it to catalytically inactive Cas13 (figure 2C). They were able to demonstrate RNA editing for programmable A to I (G) replacement of reporter, endogenous and disease-associated transcripts. However, many off-target events were observed adjacent to the guide RNAs. The investigators then determined that the ADAR2 motif of their fusion protein was responsible for the off-target mutations and performed rationale mutagenesis to enhance specificity to the intended target site. Editing nucleic acid sequence with this tool offers several advantages including the lack of a prerequisite sequence constraint such as a PAM, activity only on transcribed sequences, direct deamination without requiring endogenous repair pathways and, lastly, the transient nature of RNA rather than DNA modification.
Activation and repression of transcription with Cas9
The Cas9 capacity to specifically target certain loci in the genome can also be employed to modify determinants of protein expression without changing nucleotide sequence. In this application, Cas9 acts as a chaperone for effector proteins, such as transcription activators, repressors or epigenetic modifiers, and guides them to specific locations. Mali et al used a nuclease-null Cas9 protein, the VP64 activation domain and promoter-specific sgRNAs to stimulate transcription.39 To maintain the targeting specificity while eliminating the possibility of creating DSBs the authors generated nuclease-dead Cas9 variants by mutating the RuvC and HNH nuclease domains. To localise a transcription-activating domain to the Cas9/sgRNA complexed to complementary DNA, they then fused the VP64 activation domain to either the C terminus of nuclease-deficient Cas9 or to the specific sgRNAs. They were able to successfully activate transcription of ZFP42 (REX1) and POU5F1 (OCT4) in HEK 293 T cells by combining the nuclease-null Cas9 fused to VP64 with sgRNA specific to a 5 kb stretch of DNA proximal to the respective transcription start sites. They demonstrated similar results by fusing VP64 to sgRNA in the presence of Cas9. The inclusion of multiple synergistic sgRNAs further amplified transcription. However, the authors designed and validated a reporter assay which demonstrated there is considerable variability in off-target specificity among sgRNAs. Similar strategies using TALENs40 and ZFNs have been used, including using a ZFN-VP64 fusion to promote transcription of the gamma globin gene in a human erythroleukaemia cell line K562.41
Epigenetic modification status also determines the transcriptional activity of genes. Hilton et al described yet another application of fusing the specificity of nuclease-deficient Cas9 with sgRNA to a protein effector, in this instance the catalytic core of human acetyltransferase p300 to catalyse the acetylation of histone H3 lysine 37.42 They observed transcriptional activation associated with targeted p300 epigenetic modification of enhancers both proximal and distal to the intended gene. Similar fusions of other epigenetic modifiers including demethylases, methyltransferases and deacetylases have been generated and are likely to greatly expand the capability to perform complex and precise epigenetic modification.43–47
Delivery of effectors for genome editing
To achieve successful targeted gene therapy, efficient delivery vehicles and vectors are essential. Plasmids encoding proteins responsible for genome editing must successfully enter the nucleus of targeted cells in order to facilitate transcription and eventually translation.48 The cell and nuclear membrane are physical barriers to the passage of large hydrophilic molecules like DNA, RNA and proteins. Furthermore, DNA, RNA and protein are all subject to intracellular and extracellular degradation by nucleases and proteases.49 Methods for circumventing the physical and degradative barriers to delivery can generally be divided into viral vectors, non-viral vectors, non-vector agents and cellular delivery vehicles.
Viral vectors
Retroviruses, adenoviruses and adeno-associated viruses (AAV) are the three primary classes of viruses that have been used to deliver genetic material50 and can potentially be used in vitro, ex vivo and in vivo. Retroviral vectors use reverse transcription for replication and a subtype, lentiviruses, can integrate viral DNA into cells without the need for replication. Retroviruses have successfully edited DNA in gene therapy trials; however, the risk of oncogenesis cannot be understated.51 52 More recently lentiviruses have been modified to contain weaker cellular promoters, termed self-inactivating, and carry much reduced risk of mutagenesis.53 Integrase-defective lentiviral vectors do not integrate into the host genome, remain episomal and gradually dilute via cell division, and have been used for transient expression of ZFNs and donor templates in vitro.54
Adenoviral vectors deliver viral double-stranded DNA to the nucleus, allowing transient expression of the desired proteins such as nucleases. AAV vectors are smaller viral particles that rarely integrate into the host genome.55 They have garnered considerable attention for their ability to deliver small nucleases, such as ZFNs or Cas9, to sites of interest. An AAV vector was used to correct murine models of haemophilia A and B via a donor template and ZFN to induce liver-specific human factor VIII and IX within the albumin gene.56 Despite numerous preclinical and clinical successes, there is still a considerable lack of knowledge about the long-term efficacy and safety of AAVs as it pertains to gene editing for human disease. AAVs also suffer from size limits for the expressed transgenes, pre-existing immunity against AAV vectors as well as humoral responses, potential for limited durability in dividing cells, genomic integration’s association with mutagenesis and lack of precise control of the therapeutic gene.57 58 Indeed, Cox et al had to modify and reduce their Cas13-ADAR2 fusion construct to fit within the 4.7 kb size limit of an AAV for delivery to HEK293T.38
Non-viral vectors
In general, non-viral delivery vectors include nanoparticles and cationic carriers. Nanoparticles may comprise genomic material complexed with cations, allowing endocytosis and cell membrane transfer of negatively charged DNA. Cationic nucleic acid carriers can be divided into lipid-based agents, such as lipofectamine, and polymeric.59 These non-viral vectors offer several advantages including transient expression, the capacity for repeated administration, potential for larger strand genomic delivery and possibly improved efficacy.60 Lipofectamine has been used for successful delivery both in vitro and in vivo, as well as for CRISPR/Cas9-mediated modification of murine iPSCs.61 However, in general, these agents have to date not been used extensively in clinical application.
Cellular vectors
Gene editing can also be performed on cells ex vivo. After return to the host, gene-edited cells may elicit long-lasting biologic effects. Chimeric antigen receptor (CAR) T cells have had remarkable success in treatment of otherwise lethal haematological malignancies and have been shown to expand and persist during and after treatment.62 Briefly, T cells are isolated from human subjects by leukapheresis and gene editing performed ex vivo to induce expression of an extracellular single-chain antigen-binding domain (scFv) fused to CD137-CD3z signalling domains.63 The resultant modified CAR T cells are then reinfused to the host for targeted effector function. T lymphocytes have tremendous capacity to drive immune response—both adaptive and pathologic—in an antigen-specific manner. Gene therapies that seek to capitalise on the narrow specificity and cytotoxic potential of T cells are under study in a number of autoimmune diseases.
Non-vector delivery methods
Electroporation is historically the most widely recognised and used method of facilitating the delivery of genomic material into cells and subsequently the nucleus. It has been used for ex vivo genome editing by introduction of Cas9 and sgRNA-encoding plasmids into haematopoietic stem and progenitor cells as well as T cells.64 Mechanical deformation of the cell with microfluidic devices has also been used for efficient CRISPR-mediated gene editing.65 Finally, direct injection of targeted cells or tissues has been used to bypass the cell and nuclear membranes. Hydrodynamic injection induces tissue damage but has been shown to be effective.66 Generation of experimental animals is also accomplished in a one-step fashion with injection of genome-editing machinery into embryos or zygotes. Microinjection of Cas9 mRNA and sgRNAs into single-cell mouse embryos can effectively target multiple genes.67 68
Future directions for delivery methods
Viral vector, non-viral vector and non-vector approaches for delivery of the molecular machinery responsible for genetic editing all remain foci of active research, especially in regard to increasing cell or tissue-specific delivery. One specificity-enhancing strategy is exemplified by modification of the CRISPR-Cas endonuclease to include asialoglycoprotein receptor ligands that are preferentially internalised into hepatocytes.69
Targets for gene editing in inflammatory diseases
Chimeric autoantibody receptor T cells
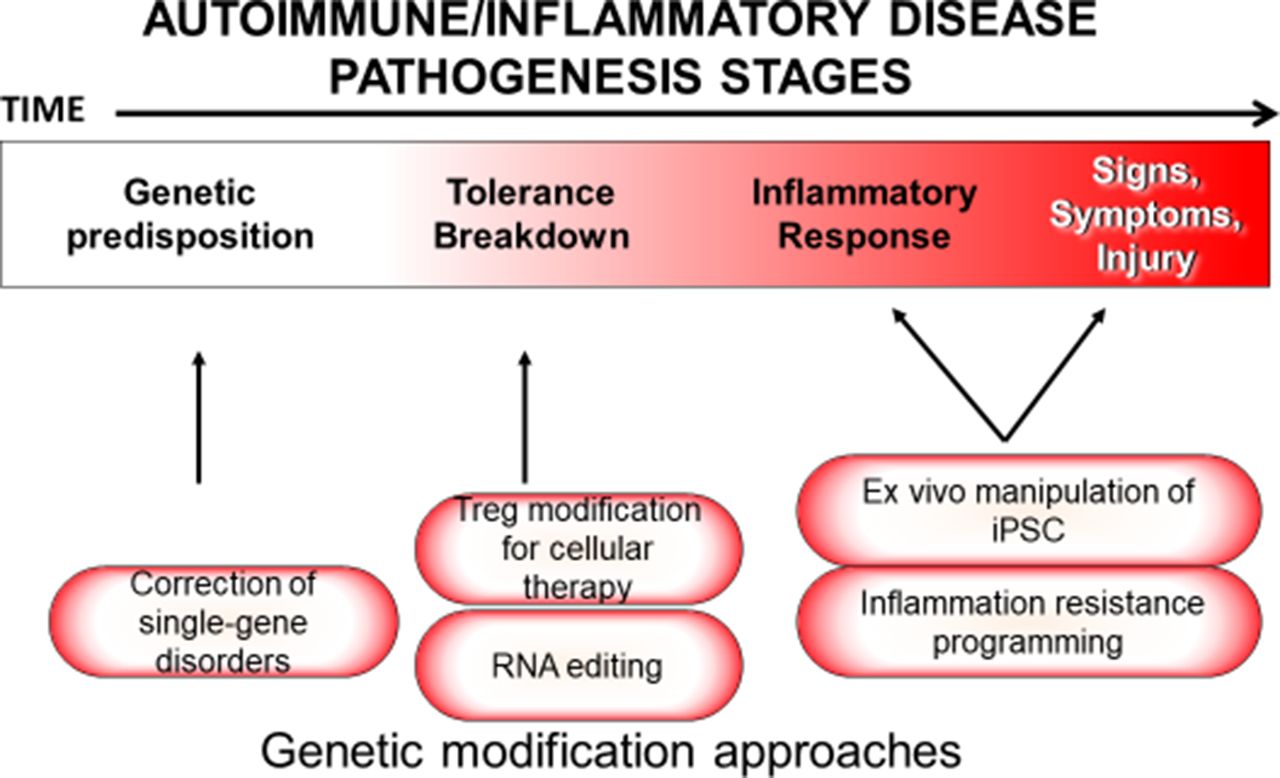
Due to the non-specific nature of current immunosuppressive treatment protocols, there is considerable interest in disrupting pathologic components of the immune system while otherwise preserving its natural function (figure 3).

Pathogenetic stages and methods by which inflammatory disease could be treated by gene editing at each stage. iPSC, induced pluripotent stem cell; Treg, T regulatory cell.
Ellebrecht et al reported a murine proof of concept using modified T cells for the treatment of the autoimmune skin disease pemphigus vulgaris (PV).70 The authors reasoned that T cells could be genetically modified to specifically recognise and eliminate pathogenic B cell clones that express autoreactive antigen receptors while maintaining the rest of the B cell compartment. The approach of targeting individual B cell clones differs from conventional anti-CD20 therapies, which deplete the entire CD20 B cell compartment. They studied PV because epitopes targeted by pathologic antibodies are well described. To specifically target the pathologic autoantibody-producing B cells, they used a lentiviral vector to genetically modify T cells to express autoantigen desmoglein (Dsg) 3 fused to CD137-CD3z signalling domains. Resulting chimeric autoantibody receptor (CAAR) T cells were then infused to treat animal models of PV, including a humanised mouse model of disease. CAAR T cells were found to expand, persist and exhibit cytotoxicity against cells expressing anti-Dsg3 B cell receptors in diseased recipients in vivo. Histopathologically, the CAAR T cell-treated mice showed an absence of IgG deposition in mucosal samples and no histologic blister formation. CAAR T cells were able to infiltrate the epidermis in a human xenograft model and similarly ameliorated the disease process. Importantly, cytotoxicity occurred without apparent off-target toxic effects.
Treatment of monogenic inflammatory diseases
The genetic characterisation of inherited periodic fever syndromes has progressed remarkably in the era of rapid, inexpensive genome sequencing. Causative single mutations have been identified in a number of heritable syndromes, raising the possibility of corrective genetic therapy for these disorders. Familial Mediterranean fever (FMF) is caused by mutations in the MEFV gene.71 Cryopyrin-associated periodic syndrome (CAPS), which encompasses familial cold autoinflammatory syndrome, Muckle-Wells syndrome and neonatal-onset multisystem inflammatory disease, is associated with mutations in the NLRP3 gene, which encodes cryopyrin, a component of the inflammasome, on chromosome 1q44.72 73 Several other examples of hereditary autoinflammatory diseases include: hyperimmunoglobulinaemia D with recurrent fevers,74 tumour necrosis factor (TNF) receptor-associated periodic syndrome (TRAPS),75 Blau syndrome,76 pyogenic arthritis, pyoderma gangrenosum and acne syndrome (PAPA),77 and chronic recurrent multifocal osteomyelitis.78 There are also increasingly well-described examples of monogenic diseases that, among other pathologies, display a phenotype of severe derangement of immunity leading to autoimmunity: Aicardi-Goutières syndrome,79 STING-associated vasculopathy with onset in infancy (SAVI), chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE)80 and many other immunodeficient conditions.81
Wiskott-Aldrich syndrome (WAS) is an example of monogenic autoimmunity-associated disorder that has been approached with gene therapy. WAS is a rare X linked immunodeficiency caused by coding variants in the WAS gene, the protein product of which regulates the actin cytoskeleton in haematopoietic lineages. Patients have thrombocytopenia, recurrent infections, eczema, an increased incidence of autoimmunity, higher risk of lymphoproliferative disorders and lymphoid malignancies, and frequently die during the third decade of life.82 83 HLA-matched allogenic haematopoietic stem cell transplantation (HSCT) is curative; however, significant risks of procedure-related morbidity and mortality persist.84 In an early attempt at gene therapy for WAS using γ-retroviral vectors, 7/9 trial subjects developed acute leukaemia secondary to viral enhancer-mediated insertional mutagenesis.85 Subsequent attempts using a self-inactivating lentiviral vector for genetic modification of autologous HSCs prior to auto-HSCT have been successful, and no patients have shown leukaemic transformation to date.86 87
WAS is not the only human condition in which enthusiasm for genetic modification approaches has been tempered by safety concerns. Attempted treatment of severe-combined immunodeficiency was marred by leukaemogenic transformation secondary to retroviral insertion of enhancers of oncogenic genes.52 More recently, use of an AAV for in vivo transduction of spinal alpha motor neurons in non-human primates and piglets with a human SMN transgene resulted in severe hepatitis and degeneration of the targeted tissues.88 These outcomes suggested an ongoing need for careful preclinical and clinical safety studies when manipulating the genome for therapies of human disease. This is especially true for many of the monogenic autoinflammatory diseases which pose lesser morbidity and mortality threats compared with severe syndromes such as WAS.
In some autoinflammatory syndromes, such as FMF, CAPS and TRAPS, the pathogenic cell type is likely to be bone marrow derived and radio and/or chemotherapy sensitive. These conditions thus represent ideal first targets or ‘low-hanging fruit’ for cure or amelioration with genetic modification of haematopoietic cells. In contrast, tissue injury from interferonopathies such as SAVI and CANDLE is likely to be caused by complex multicellular gene-phenotype interactions, and thus strategies targeting multiple cell types and tissues will be required. While still their infancy, highly efficient base editing techniques (see section above) may make genetic editing for both lethal and non-lethal monogenic disease a tenable strategy.
Stem cell modification
There is considerable interest in genetically modifying iPSCs for the treatment of human disease. Advantages of iPSCs include their potential durability in vivo, their ability to differentiate into tissues affected by chronic inflammatory diseases and the ability to create abundant quantities without the use of embryos or other products of conception.
Controlled delivery of anticytokine therapy
One approach for the use of genetically modified iPSCs is known as closed-loop biologic drug delivery system. Anticytokine therapies with monoclonal antibodies and decoy receptors have revolutionised the treatment of chronic inflammatory diseases in the last two decades.89 However, targeting key proinflammatory molecules such as TNF-α or interleukins (IL) entails a risk of side effects, including infections, and requires constant dosing and patient exposure to the drug which may interfere with the pleiotropic roles of the targeted cytokine.90 To address the problem of non-specific loss of cytokine function in all tissues created by such therapies, Brunger et al sought to engineer resistance to cytokine function only among cells of interest. They modified murine iPSCs using CRISPR/Cas9 to insert anti-inflammatory molecules (eg, IL-1Ra or chimeric human sTNFR1-murine IgG) in the Ccl2 locus.91 The Ccl2 gene product regulates trafficking of inflammatory monocytes/macrophages, basophils and T lymphocytes in response to inflammatory cues such as TNF-α and IL-1.92 The inserted naturally occurring cytokine antagonists within the Ccl2 locus mitigated the inflammatory effects of physiologic concentrations of IL-1 and TNF-α when the iPSCs were cultured in monolayer as well as after differentiation into engineered cartilage tissues. The engineered cartilage tissues were observed to be resistant to the pathophysiologic effects of IL-1 and TNF-α. The authors proposed that this approach could be used for a targeted cell-based anticytokine vaccine.
Modification of stems cells for tissue regeneration
Approaches utilising genetic modifications have found recent application in the field of regenerative medicine in attempts to restore function of damaged and diseased tissues.93–96
Osteoarthritis (OA) is a highly prevalent disabling disease for which disease-modifying therapy (other than joint replacement) has not been identified. However, OA joints are characterised by the presence of proinflammatory cytokines,97 98 which could hinder the therapeutic effect, engraftment and/or longevity of stem cell-based therapy in that milieu. Rather than utilising genetically engineered iPSCs to modify inflammatory signalling at the local tissue level, Brunger et al used CRISPR/Cas9 to engineer murine iPSCs to be inflammation resistant61 with the aim of utilising the engineered cells for regenerative medicine in a hostile, inflammatory microenvironment. They deleted the IL-1 receptor type 1 gene, selected clones with homozygous deletion and used specific differentiating factors to generate cartilage from the edited iPSC clones (figure 4). The resulting genetically modified cartilage was resistant to cytokine-mediated tissue degradation relative to wild-type and heterozygous clone cartilage.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic illustration of the strategy for generating iPSCs resistant to IL-1-mediated signalling for tissue engineering applications. (A) Binding of IL-1 ligand to the IL-1RI results in activation of a proinflammatory transcription programme involving the transcription factors NF-κB, JNK and MSK-1. (B) gRNAs target the genome-editing nuclease Cas9 to two sites flanking exon 2 of IL-1RI, which encodes the signal peptide sequence. (C) Cas9 induces DNA DSBs, which may be repaired via NHEJ. (D) NHEJ leads to a subset of alleles with fully intact IL-1RI, while others may have genomic disruptions at the IL-1RI locus, including excision of the signal peptide sequence, resulting in loss of signalling through IL-1RI. DSB, double-stranded break; gRNA, guide RNA; IL-1, interleukin-1; IL-1R1, IL-1 receptor type 1; iPSC, induced pluripotent stem cell; NHEJ, non-homologous end joining. (Reproduced from Brunger et al 61 with permission; © Arthritis and Rheumatology)
Webber et al also used iPSCs to explore the possibility of tissue regeneration in recessive dystrophic epidermolysis bullosa, a severe disorder caused by mutations to the COL7A1 gene that causes life-threatening derangement of skin integrity.99 Patient-derived primary fibroblasts were isolated and the COL7A1 gene defect restored to wild-type status using CRISPR/Cas9. Genetic modification was assessed and found to be specific to the intended locus apart from a single, predicted off-target modification in a clone treated with Cas9 nuclease rather than the nickase. iPSCs were generated from the modified fibroblasts and showed ability to differentiate into keratinocytes, mesenchymal stem cells and HSCs.
T regulatory cells
T regulatory cells (Tregs), defined classically as CD4+CD25+FOXP3+, are a naturally occurring subset of helper T cells that have immunoregulatory functions including suppression of antigen-specific T cells and maintenance of peripheral tolerance. The observed frequency in suppressive function of Tregs in association with autoimmune disease100 101 has led to a growing interest in their potential therapeutic use. Effectiveness of Treg cellular therapy depends on availability of large numbers of Tregs—many times greater than the quantities of naturally occurring Tregs available for harvest. In vitro expansion protocols are therefore essential to Treg cellular therapeutic approaches. However, there are barriers to efficient in vitro enrichment of antigen-specific T cells,102 such as loss of regulatory phenotype and insufficient in vitro expansion. These obstacles may be overcome with gene editing techniques. There is also interest in using gene editing to induce in vivo production of antigen-specific Tregs. The use of gene editing of Tregs has recently been comprehensively reviewed.103
Genetically modified Treg cell therapy
Isolating sufficient numbers of Tregs with or without a rare antigenic specificity from the natural T cell population is a major challenge for Treg-focused therapy. Several recent strategies for producing large quantities of clinical grade Tregs and for generating antigen-specific Tregs in vitro have employed genome engineering. CD4+ T cells have been modified ex vivo to express the ‘master’ transcription factor FOXP3 in a fashion that allows them to differentiate into a large population of Tregs. Treg populations expanded by inducible FoxP3 have been used to treat immunodysregulation polyendocrinopathy enteropathy X linked syndrome, caused by a dysfunction in the FoxP3 gene, in human patients as well as animal models of autoimmunity.104–106
Another approach is the use of TCR gene transfer to direct polyclonal Tregs to express a specific TCR gene and thus redirect specificity towards a single antigenic epitope. This technique has been demonstrated to be effective in several murine models of transplant tolerance, inflammatory arthritis and in human T cells for islet cell targeting.107–110
Finally, in a manner analogous to the generation of CAR T cells for therapy of malignancy, Tregs can be engineered to express extracellular scFv or antigenic domains fused to downstream intracellular signalling molecules. Antigen-specific chimeric receptors have been used in murine experimental autoimmune encephalitis (EAE)111 112 and a murine model of inflammatory bowel disease.113 114 There are ongoing efforts to examine the utility of citrullinated-peptide-specific CAR Tregs for the treatment of rheumatoid arthritis.115 Chimeric autoantigen receptor Tregs have been studied in a murine model of haemophilia A with autoantibody formation to factor VIII.116 Autoantibody-producing B cells were able to be ‘tolerized’ and autoantibody production halted.
Induction of antigen-specific Tregs in vivo
The liver is a robust reticuloendothelial organ and contains resident cells that support a tolerogenic effect on self and foreign antigens by expression of surface ligand inhibitors for T cell activation and production of inflammatory mediators.117–119 As such it is uniquely positioned as a target for genetic therapies for induction of antigen-specific tolerance. Keeler et al used an AAV8 vector with the cDNA for the neuroprotein, myelin oligodendrocyte glycoprotein (MOG), under the control of a liver-specific promoter to induce hepatic expression of MOG in a murine model of multiple sclerosis, EAE. Hepatic expression of MOG used the tolerogenic properties of the liver to produce MOG-specific FOXP3+ Tregs in vivo. Mice treated prophylactically were protected from developing EAE and in mice already experiencing mild to moderate neurological deficits the vector alone was effective at reversing clinical and pathological signs of disease. When combined with immunosuppression, AAV immunotherapy rescued mice from fatal end-stage EAE and severe paralysis.120 Using a similar strategy, a lentiviral vector was used to induce insulin B chain 9-23, the immunodominant T cell epitope in non-obese diabetic (NOD) mice, in NOD hepatocytes. The lentiviral treatment induced insulin B chain 9-23-specific effector T cells but also FoxP3+ Tregs, which halted islet immune cell infiltration and protected from T1D. When combined with anti-CD3 monoclonal antibody, T1D was reversed, and shown to be dependent on Tregs.121
RNA editing and transcriptomic modification
The technology for and application of RNA editing is still in its infancy relative to the technologies for making permanent changes to the genome. However, there are speculative applications of RNA editing for inflammatory diseases. Cox et al were able to repair transcripts of the 878G>A (AVPR2 W293X) mutation associated with X linked nephrogenic diabetes insipidus and the 1517G>A (FANCC W506X) mutation associated with Fanconi anaemia.38 They also suggested the possibility of utilising similar methods for correction of any of the thousands of G to A mutations associated with pathologic states, potentially offering the ability to mimic protective alleles for at-risk individuals during times of immunologic and inflammatory stress.
Conclusions
The technology to manipulate genomic material has allowed tremendous discovery in the biology of inflammatory disease. While still in its infancy, genetic editing is now, decades after the discovery of the nature and structure of DNA, entering the realm of therapy (table 2). This is particularly true for severe monogenic disease. There are several apparent niches for gene editing in the treatment of inflammatory diseases, including correction of monogenic autoinflammatory syndromes, CAAR T cell therapy for autoantigen-specific targeting of pathologic B cell clones, modification of iPSCs for controlled cytokine delivery and tissue regeneration and Treg-based therapies. As technology matures, cell therapies based in genome editing will likely strive towards the ‘holy grail’ of autoimmune disease therapy, that is, targeted inhibition of the pathologic components of the immune system without necessitating generalised immunosuppression. New tools for transient modification of protein structure, function and expression will allow us to fine-tune the delicate balance the immune system maintains between defence and tolerance to self. Harnessing the inherent specificity of our own immune system to target pathologic B or T cell clones, particular cellular subsets necessary for disease or disrupt trafficking to affected tissues will likely become feasible in the near future. Specific therapy will more potently inhibit the inflammatory processes yet spare the remainder of the immune system. But therapies utilising genetic editing will likely expand beyond ex vivo modification of cells of the immune system when we can reliably target certain cell types or tissues in vivo. One can imagine stepwise approaches for targeted genetic honing to redirect from inflammation to homeostasis in chronic inflammatory diseases affecting a particular organ or cell type. Addition to or antagonism of various growth factors targeted to organ parenchymal or interstitial cells could potentially be applied to fibrotic inflammatory diseases, which at present have a paucity of treatment options. Furthermore, more elegant and coordinated control of gene expression could allow the capability to generate replacements for organs or tissues damaged by inflammation, such as a biologic rather than plastic and metallic replacement for a joint with end-stage OA. The increasing capability, ease of use and reliability of modern tools for gene editing will undoubtedly lead to new pathophysiologic insights and therapies for immune and inflammatory diseases.
Human inflammatory disease states for which gene editing approaches and therapies have been considered, and for which studies in animal models or primary human cells have been reported
References
Footnotes
Handling editor Josef S Smolen
Contributors All authors contributed to the conception and design as well as the literature review for this work. Each participated in drafting and revising the important intellectual content and approved the final version to be published. All are accountable for all aspects of this work.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Not required.
Provenance and peer review Commissioned; externally peer reviewed.