Article Text

Abstract
Introduction Inflammatory joint diseases such as rheumatoid arthritis are associated with local bone erosions and systemic bone loss, mediated by increased osteoclastic activity. The receptor activator of nuclear factor (NF) κB ligand (RANKL) plays a key role in mediating inflammation-induced bone loss, whereas tumour necrosis factor (TNF) plays a central role in the inflammatory process. Here we tested whether a recently identified class of small molecule inhibitors of RANKL signalling (ABD compounds) also affect TNF signalling and whether these compounds inhibit inflammation in an animal model of rheumatoid arthritis.
Methods The inhibitory effects of the ABD compounds on TNF-induced signalling were tested in mouse macrophage cultures by western blotting and in an NFκB luciferase-reporter cell line. The anti-inflammatory effects of the compounds were tested in the mouse collagen-induced arthritis model of rheumatoid arthritis.
Results The ABD compounds ABD328 and ABD345 both inhibited TNF-induced activation of the NFκB pathway and the extracellular signal-regulated kinase (ERK) and Jun kinase (JNK) mitogen activated protein kinases (MAPKs). When tested in the mouse collagen-induced arthritis model of rheumatoid arthritis, the compounds suppressed inflammatory arthritis, inhibited joint destruction and prevented systemic bone loss. Furthermore, one of the compounds (ABD328) showed oral activity.
Conclusions Here we describe a novel class of small molecule compounds that inhibit both RANKL- and TNF-induced NFκB and MAPK signalling in osteoclasts and macrophages, and inflammation and bone destruction in a mouse model of rheumatoid arthritis. These novel compounds therefore represent a promising new class of treatments for inflammatory diseases, such as rheumatoid arthritis.
- Rheumatoid Arthritis
- TNF-alpha
- Osteoporosis
Statistics from Altmetric.com
Introduction
Increased osteoclastic bone resorption plays a major role in the pathogenesis of several diseases such as osteoporosis, rheumatoid arthritis and periodontal disease. Receptor activator of nuclear factor (NF)-κB (RANK), a member of the tumour necrosis factor (TNF) receptor superfamily (TNFRSF), plays a central role in regulating osteoclast differentiation and function.1 When RANK is activated by receptor activator of nuclear factor κB ligand (RANKL), a signalling complex is recruited to the intracellular domain of the receptor and this triggers activation of several downstream signalling pathways, such as NFκB and mitogen activated protein kinase (MAPK) pathways, which promote osteoclast differentiation and bone resorption.2 Pro-inflammatory cytokines such as TNF also cause NFκB and MAPK activation. While activation of the RANK signalling pathway plays a critical role in mediating the osteoclastic bone destruction that accompanies inflammatory arthritis, RANK is not involved in the pathogenesis of joint inflammation, since osteoprotegerin—a decoy receptor for RANKL—inhibits bone destruction, but not joint inflammation in models of inflammatory arthritis.3 While TNF plays a critical role in the pathogenesis of inflammation and enhances RANKL-induced osteoclast formation in vitro,4 TNF-dependent arthritis is not accompanied by bone destruction in Fos−/− mice which lack osteoclasts.5 Moreover, intervention studies have shown that a combination of treatments which block RANKL-induced osteoclast activation and TNF-induced inflammatory signalling is necessary to fully suppress inflammation and to prevent bone destruction in arthritis.6 These findings illustrate that RANKL-induced osteoclast activation and TNF-induced pro-inflammatory signalling play differing and complementary roles as mediators of inflammation and bone destruction in inflammatory diseases. Although peptide inhibitors of NFκB signalling have been shown to suppress inflammation and bone destruction in models of arthritis,7 we are unaware of any small molecules which are capable of inhibiting signalling downstream of both the TNF and RANK receptors. We previously identified a series of biphenylcarboxylic acid derivatives which were found to inhibit osteoclastic bone resorption in vitro and ovariectomy-induced bone loss in vivo by causing osteoclast apoptosis.8 Previous studies showed that the parent derivative, the butanediol biphenylcarboxylic acid ester (ABD56), acted by inhibiting RANKL-induced phosphorylation of IκB and extracellular signal-regulated kinase (ERK).9 Although active, ABD56 is not metabolically stable as the ester bond is easily cleaved. We therefore developed more stable compounds where the ester bond was replaced by a ketone. The stabilised derivatives ABD328 and ABD345 showed higher potency in osteoclast cultures and ABD328 showed oral activity in the ovx mouse model of osteoporosis.10 The receptors for TNF and RANK (the receptor for RANKL) are both members of the TNFRSF, and activate signalling through recruitment of TNF-receptor associated factor (TRAF) molecules.11 ,12 We therefore studied whether ABD328 and ABD345 inhibit TNF-induced NFκB and MAPK activation and inflammation and bone loss in the collagen-induced arthritis (CIA) mouse model of RA.
Materials and methods
Materials
Chemical reagents were obtained from Sigma (Dorset, UK) and culture media were from Gibco (UK). The test compounds ABD328 and ABD345 were synthesised inhouse using standard methods and their identity and purity were confirmed by combustion analysis, mass spectrometry and nuclear magnetic resonance (NMR) as previously described.13 ,14 Recombinant human RANKL was produced as described by Idris et al.15 All other cytokines were obtained from R&D Systems (Abingdon, UK).
Cell culture
Osteoclasts and bone marrow macrophages were generated from bone marrow cells obtained from the long bones of 3–5-month-old mice as previously described.16 For generation of bone marrow macrophages, the cells were cultured in αminimal essential medium (MEM) supplemented with 10% foetal calf serum (FCS), antibiotics and recombinant mouse macrophage colony stimulating factor (M-CSF) (100 ng/mL) for 3 days, the non-adherent cells removed and the macrophages harvested using cell dissociation buffer (Gibco) and scraping and replated at 104 cells per well in 96-well plates. Macrophage viability was measured using the Alamar Blue assay as described previously.16 For generation of osteoclasts, the M-CSF treated bone marrow macrophages were plated in 96-well plates (104 cells/well) in 150 μL of αMEM supplemented with 10% FCS, antibiotics, M-CSF (25 ng/mL) and RANKL (100 ng/mL).
Intracellular signalling
Bone marrow macrophages were generated as described above and plated in 6-well plates at 3×105 cells per well in 2 mL of αMEM supplemented with 10% FCS, antibiotics and recombinant mouse M-CSF (100 ng/mL). When the cultures reached semiconfluence, the medium was replaced with serum-free medium and the cells serum starved for 2 h. Next, test compounds or vehicle (0.1% DMSO) were added to the media, and the cells cultured for a further 1 h. Cells were stimulated with cytokines for the amount of time indicated, washed in phosphate buffered saline (PBS) and homogenised in lysis buffer (PBS with 0.1% (w/v) sodium dodecyl sulfate (SDS), 0.5% (w/v) sodium deoxycholate and 2% (v/v) protease inhibitor cocktail). The cell lysates were centrifuged at 14 000 g for 10 min at 4°C, the supernatants collected and protein concentrations determined using a Bio-Rad protein assay kit. Aliquots of the cell lysates containing 50 µg protein per lane were run on a 10% SDS-acrylamide gel and blotted onto a nylon membrane. The membranes were washed in Tris buffered saline (TBS) and incubated overnight at 4°C with the relevant primary antibodies (1 : 1000 dilution): actin (Sigma, Poole, UK), phospho-IκBa, total IκBa, phospho-p42/44, total p42/44, phospho-jnk, phospho-p38, phospho-STAT1 and phospho-STAT3 (Cell Signalling Technology, USA). Subsequently, the membranes were washed in TBS and incubated with the appropriate secondary antibodies coupled to horse radish peroxidase (HRP), washed in TBS again, and bands visualised using chemiluminescence (Amersham, UK) on a Syngene GeneGnome imaging system.
Luciferase reporter assays
The 293/NFκB-luc cell line (Cambridge Scientific, UK) was cultured at 104 cells per well in black 96-well plates in 100 µL D-MEM supplemented with 10% FCS and antibiotics for 24 h. Next the medium was replaced with serum free medium with or without test compounds, and the cells cultured for a further 1 h. The cells were stimulated with TNFα (R&D, 10 ng/mL) for 3 h and luciferase activity measured using the Promega Steady-Glo system on a Biotek HT platereader.
Collagen-induced arthritis
Male DBA/1 mice (8-week old; Harlan, UK) were given a 100 µL intradermal injection of an emulsion of chicken collagen type II (Sigma, 2 mg/mL) in complete Freund's adjuvant supplemented with 1 mg/mL freeze dried Mycobacterium tuberculosis (Difco) at the base of the tail. Inflammation of the paws was generally first observed after approximately 2 weeks. At this stage, treatment was started by either daily gavage or intraperitoneal injection of drug (10 mg/kg) or vehicle (corn oil) for 21 days. The development of joint inflammation was measured by manual scoring using the following scoring system: 0=normal, 1=erythema or swelling of one or more digits, 2=1 plus swelling of carpal joints, and 3=extensive swelling including hock joints. A total score was calculated based on the sum of counts in all four limbs.
At the end of the experiments, the animals were killed and legs dissected and fixed in phosphate buffered formaldehyde (4%) for 24 h and transferred to 70% alcohol. Paws were analysed using a Skyscan 1172 µCT system at a resolution of 17 µm (60 kV, 167 mA, 0.6° rotation step). Images were reconstructed using Skyscan NRecon software and visualised using CTVol. After µCT scanning, the samples were dehydrated in ethanol and processed to wax. The samples were cut at 7 µm using a microtome, and the sections stained for H&E. In parallel sections, osteoclasts were identified by TRAcP staining, and the sections counterstained with haematoxylin. Sections were analysed using a Zeiss Axioimager microscope fitted with a QImaging Retiga 4000 camera at 10× magnification.
For assessment of trabecular bone volume, the proximal tibiae were scanned in a Skyscan 1172 scanner at a resolution of 5 µm (60 kV, 167 mA, 0.6° rotation step). The images were reconstructed as described above and trabecular bone volume was measured in a volume of 200 slices starting 100 µm distal of the growth plate.
Statistical analysis
Statistical analysis was performed using SPSS for Windows. Between-group comparisons were by analysis of variance (ANOVA) with Dunnet's post-test.
Results
The biphenylcarboxylic acid derivatives ABD328 and ABD345 inhibit both RANKL and TNF-induced signalling
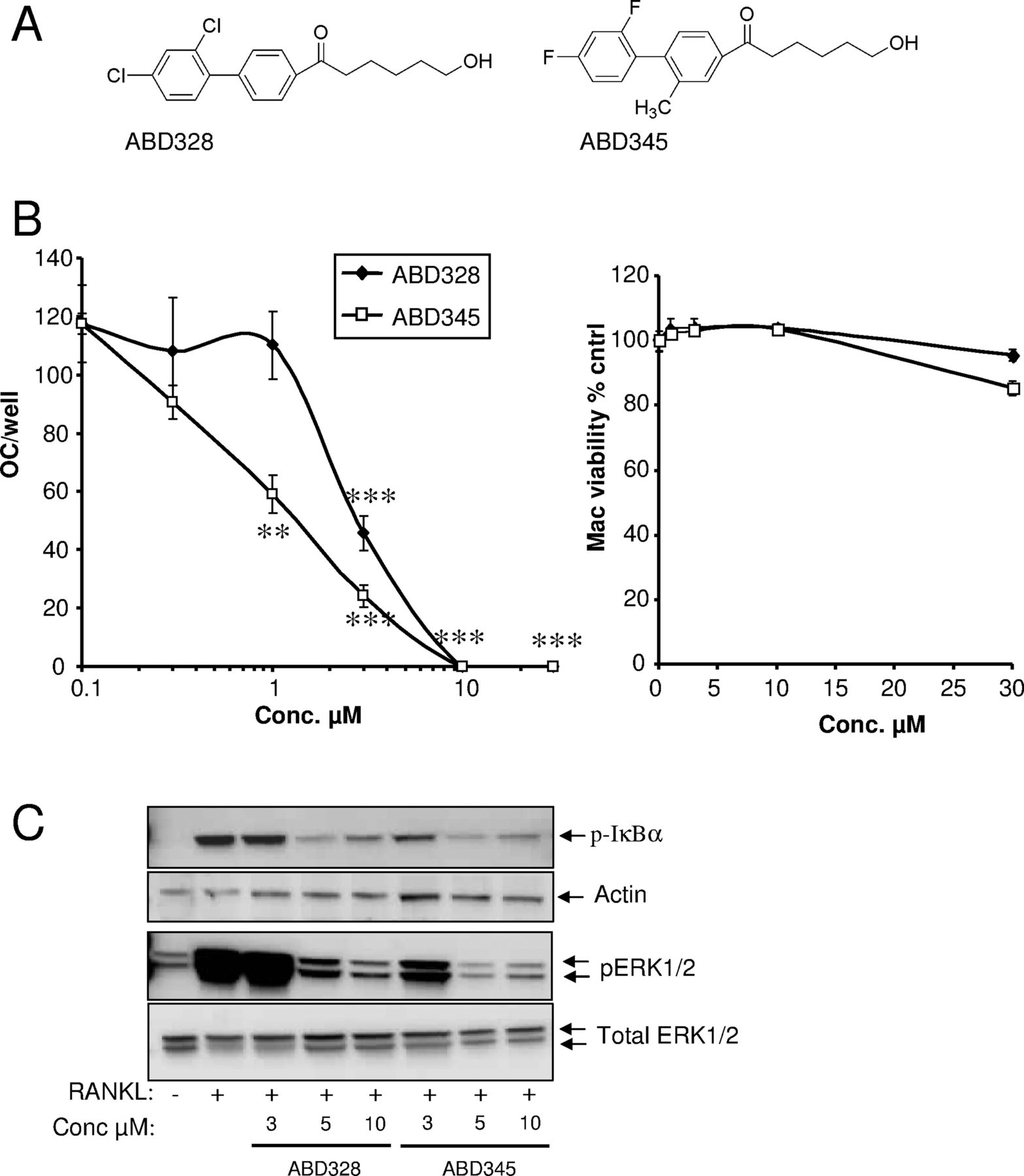
ABD328 and ABD345 were derived from the parent compound ABD56 by replacing the labile ester bond with a stable ketone linkage (figure 1A). Like the parent compound ABD56, ABD328 and ABD345 inhibited osteoclast formation in mouse bone marrow cultures without any effects on bone marrow macrophage viability (figure 1B). However, ABD328 and ABD345 were more potent than the parent compound ABD56 (IC50 8 µM8), and ABD345 (IC50 1.7±0.3 µM) was slightly more potent than ABD328 (IC50 2.2±0.2 µM). As expected, ABD328 and ABD345 inhibited RANKL-induced phosphorylation of IκB and ERK in M-CSF-dependent bone marrow macrophage cultures (figure 1C). As described above, the receptor for RANKL (RANK) is a member of the TNFR superfamily and activates many of the same signalling cascades as TNF. We therefore tested the effects of ABD328 and ABD345 on TNF-induced signalling. Some signal for phosphorylated IκB was observed in the vehicle control; however, treatment with TNF for 5 min significantly increased the phosphor-IκB signal. ABD328 and ABD345 both inhibited TNF-induced IκB and ERK phosphorylation, and appeared to inhibit TNF- and RANKL-induced signalling with equal potency (figure 2A). As in the osteoclast formation assay, ABD345 appears to be more potent than ABD328. ABD328 and ABD345 also inhibited TNF-induced phosphorylation of Jun kinase (JNK), but not of p38 MAPK (figure 2B). We next studied the effects of the compounds on activation of the downstream NFκB transcription factor by TNF using HEK293 cells stably transfected with an NFκB-luciferase reporter construct. Both ABD328 and ABD345 inhibited TNF-induced NFκB activation in this assay with ABD345 again more potent than ABD328 (figure 2C).

ABD328 and ABD345 are potent inhibitors of osteoclast formation and receptor activator of necrosis factor κB ligand (RANKL)-induced signalling. (A) Structure of the compounds. (B) Osteoclast and bone marrow macrophage cultures were treated with the compounds at the concentrations indicated. ABD328 and ABD345 inhibited osteoclast formation but had no effect on macrophage viability. The results are from a typical experiment (n=5) out of four experiments performed. **p<0.01; ***p<0.001. (C) Bone marrow macrophage cultures were serum starved and pretreated with compounds at the concentrations indicated for 1 h. The cultures were stimulated with RANKL (100 ng/mL) for 5 or 10 min and IκB (5 min) and extracellular signal-regulated kinase (ERK) (10 min) phosphorylation analysed by western blotting.

ABD compounds inhibit tumour necrosis factor (TNF)-induced signalling. (A and B) Bone marrow macrophage cultures were serum starved and pretreated with compounds at the concentrations indicated (A) or 10 µM (B) for 1 h. The cultures were stimulated with TNF (10 ng/mL) for 5 or 10 min and IκB (5 min) and extracellular signal-regulated kinase (ERK), Jun kinase (JNK) and p38 mitogen activated protein kinase (MAPK) (10 min) phosphorylation analysed by western blotting. (C) 293/NFκB-luc cells were serum starved and pretreated with compounds at the doses indicated for 1 h before stimulation with TNF (10 ng/mL) for 3 h and luciferase activity measured. The results are from a typical experiment (n=5) out of three experiments performed. **p<0.01; ***p<0.001.
ABD328 and ABD345 did not inhibit M-CSF- or IFNγ-induced signalling suggesting that the compounds specifically target the signalling complex of TNFRSF members such as RANK and the TNF receptor (see online supplementary figure S1).
ABD345 prevents collagen-induced arthritis
As ABD328 and ABD345 inhibited both RANKL- and TNF-induced signalling, these compounds could inhibit inflammation and inflammatory bone loss. As ABD345 was the more potent compound in vitro, we analysed the efficacy of this compound in the collagen-induced arthritis model of inflammatory arthritis. ABD345 was administered by daily intraperitoneal injection and showed a dose-dependent inhibition of collagen-induced arthritis (figure 3). At a dose of 10 mg/kg/day (given by an intraperitoneal injection), ABD345 almost abolished the development of collagen-induced arthritis as assessed by clinical score (figure 3A). When the paws were analysed by µCT, the vehicle control group displayed severe bone erosions near the affected joints (figure 3B), whereas these erosions were virtually absent in the ABD345 treated group. Histological analysis showed typical pannus formation with the presence of large numbers of osteoclasts and severe cartilage and bone destruction in joints from vehicle treated mice (figure 4). In keeping with the inflammation scores and µCT analysis, mice which had been treated with ABD345 at 10 mg/kg/day showed hardly any inflammatory infiltrate compared with vehicle treated mice and no cartilage or bone destruction (figure 4). At the lower dose of 5 mg/kg/day, ABD345 still showed some inhibition of arthritis; however, minor joint damage became apparent in µCT scans, and histological analysis showed some inflammatory infiltrate in some joints, although the cartilage still appeared normal, and we did not observe osteoclasts associated with the inflamed synovium. The dose of 2 mg/kg/day did not statistically significantly decrease the inflammation scores.

ABD345 dose-dependently inhibits collagen-induced arthritis. (A) Scoring of joint inflammation. Values are averages±SEM (N=10). (B) Examples of joint damage visualised by µCT analysis. Note the severe bone damage in the joints of the vehicle control specimen (yellow arrows), minor damage in the 5 mg/kg/day specimen and absence of damage in the 10 mg/kg/day sample. Data were analysed by analysis of variance (ANOVA) using the area under the curve. **p<0.01; ***p<0.001 from vehicle control.

ABD345 inhibits joint inflammation and joint destruction. Examples of histological analysis of hind paws from mice from figure 3. Note the severe joint destruction in the vehicle treated mouse with clear pannus formation (black arrow in A) associated with numerous osteoclasts (yellow arrows in B). No pannus was observed in the 10 mg/kg/day group (C and D) and cartilage appeared normal (blue arrows, C). Synovial thickening was observed in a small number of joints in the 5 mg/kg/day group (black arrow, E), but this was not associated with osteoclasts (F) and cartilage appeared normal (blue arrows, E).
ABD328 is an orally active inhibitor of collagen-induced arthritis
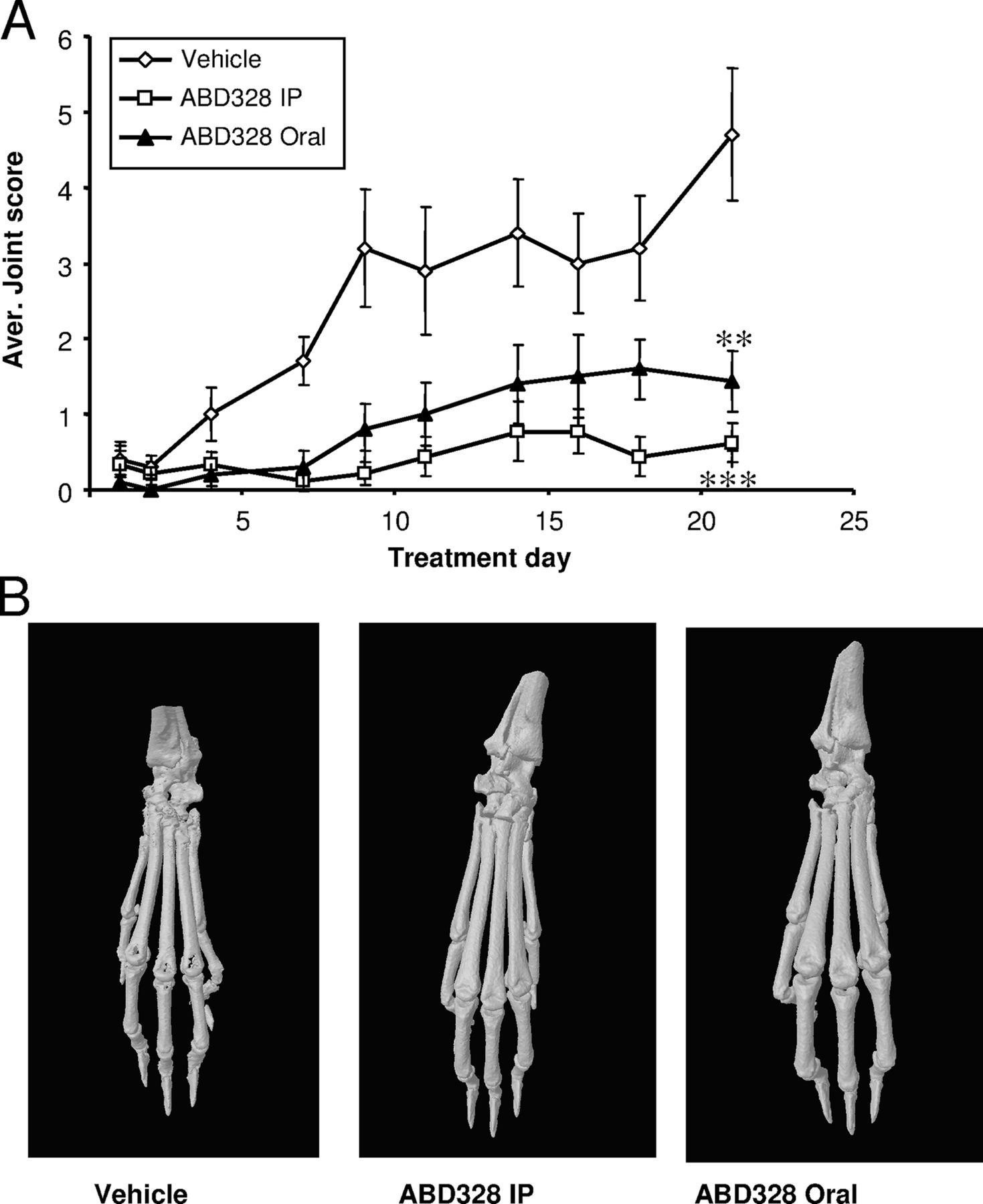
In a previous study we found that ABD328 prevents ovariectomy-induced bone loss when dosed orally using gavage,10 and we therefore tested whether this compound was orally active in the collagen induced arthritis (CIA) model as well. ABD328 given orally at 10 mg/kg/day significantly reduced progression of the inflammation scores; however, it was not as effective as the same dose administered by intraperitoneal injection (figure 5A). Histology and µCT analysis confirmed that oral ABD328 significantly reduced CIA-induced bone and cartilage destruction (figure 6).

ABD328 is an orally active inhibitor of collagen induced arthritis (CIA). (A) Scoring of joint inflammation. Values are averages±SEM (N=10). (B) Examples of joint damage visualised by µCT analysis. Note the joint damage of the vehicle control specimen and absence of damage in the samples from the ABD328 treated mice. Data were analysed by analysis of variance (ANOVA) using the area under the curve. **p<0.01; ***p<0.001 from vehicle control.

ABD328 prevents joint destruction. Examples of histological analysis of hind paws from mice from figure 5. (A and B) Vehicle control; (C and D) ABD328 10 mg/kg/day, by IP injection; (E and F) ABD328 10 mg/kg/day, by gavage. Black arrows indicate synovial thickening, yellow arrows indicate osteoclasts. Some synovial thickening was observed in ABD328 treated animals (C), but this was not associated with increased osteoclasts numbers, and cartilage appeared normal (blue arrows, C and E).
ABD328 and ABD345 inhibit inflammatory bone loss
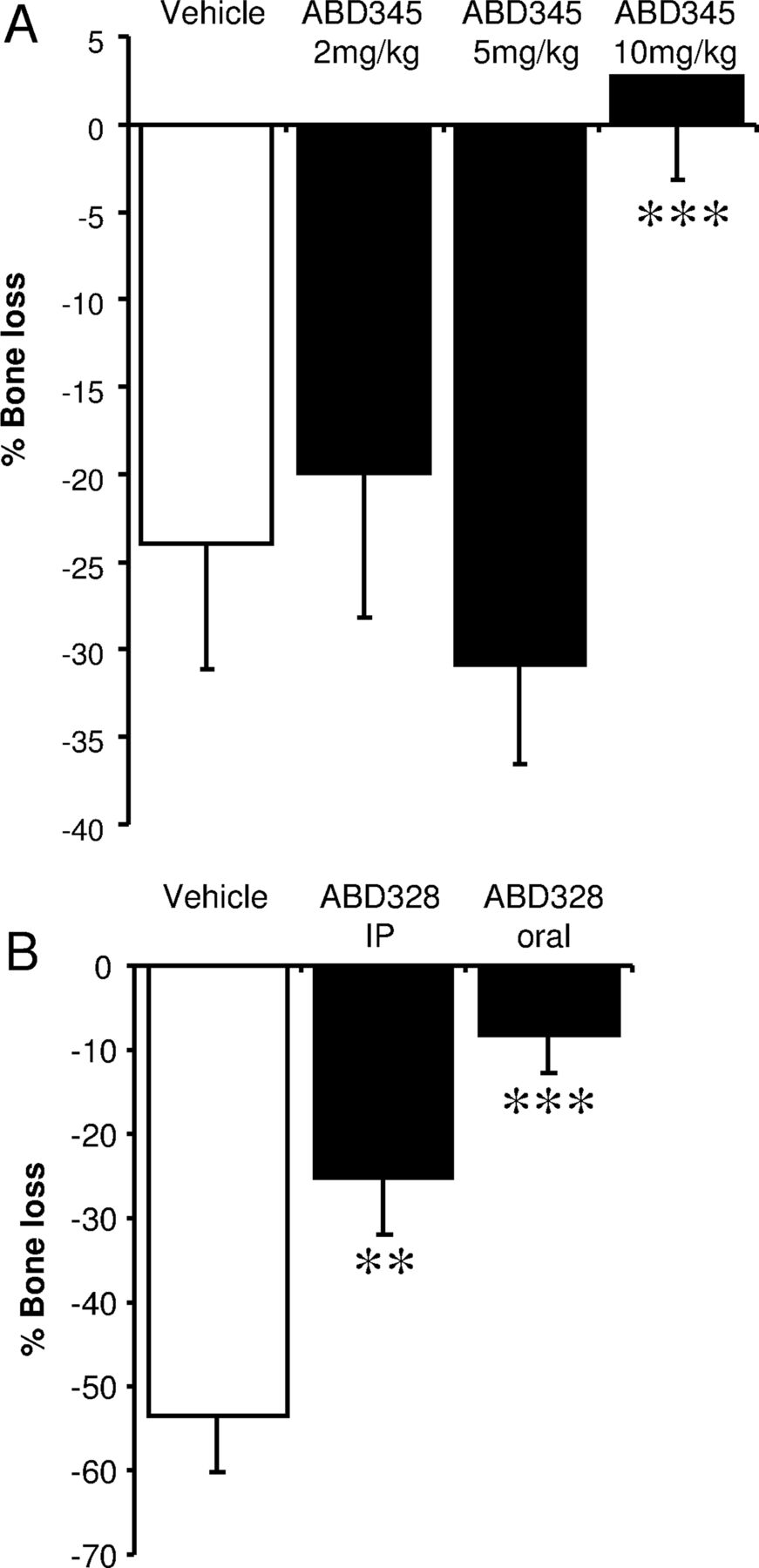
The inflammation in the CIA model leads to systemic bone loss in sites not directly affected by the inflammation, such as the knee. Figure 7A shows an almost 25% decrease in trabecular bone volume in the vehicle treated group. This bone loss is prevented by ABD345 at a dose of 10 mg/kg/day, while lower doses are not effective (figure 7A). ABD328 prevented trabecular bone loss at the same site both by intraperitoneal injection and oral dosing (figure 7B).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ABD328 and ABD345 inhibit inflammation-induced bone loss. Trabecular bone volume of the proximal tibia was measure by µCT analysis. Data are expressed as difference from control animals. collagen induced arthritis (CIA) leads to a pronounced decrease in trabecular bone volume, which is inhibited by 10 mg/kg/day ABD345 (A) and ABD328, both by intraperitoneal injection and oral dosing (B). Values are averages±SD (N=8). Data were analysed by analysis of variance (ANOVA). **p<0.01; ***p<0.001.
Discussion
In this study, we report the identification of a novel series of small molecule inhibitors of NFκB and MAPK signalling which inhibit signal transduction through both the RANK and TNF receptors. The compounds were effective at inhibiting inflammatory activity in the collagen-induced arthritis mouse model of inflammatory arthritis and the bone loss associated with this arthritis model. Although the important role of NFκB in inflammation is well known, the ERK and JNK MAPK pathways have also been shown to be activated in rheumatoid arthritis and high levels in early disease predict progression to erosive disease.17 The inhibitory effect of the compounds on TNF-induced JNK and ERK activation may therefore enhance the anti-inflammatory effects of the compounds.
The compounds did not affect macrophage viability, even at much higher concentrations than those necessary to inhibit osteoclast formation or cytokine-induced signalling. We have previously shown that these compounds also have no effect on osteoblast viability or function,16 indicating that the effects of the compounds are not due to general toxicity. In support of this, visual inspection of drug treated animals at time of death did not show any evidence of toxicity.
Although the precise molecular target for these compounds remains unclear, the evidence presented here suggests that they may inhibit NFκB and MAPK signalling by inhibiting recruitment or activation of signalling molecules common to both the TNF and RANK receptors. Signalling downstream of the TNF and RANK receptors involves the interaction of a large number of signalling molecules. One of the first steps in both RANKL and TNF signalling is recruitment of TRAF molecules to the RANK and TNF receptors. However, TNF-induced signalling is mostly dependent on TRAF-2 and -5, whereas RANKL-induced signalling is mostly mediated by TRAF-6. As the binding motif for TRAF-6 is different to that for TRAF-2 and TRAF-5,11 ,18 it is unlikely that the compounds directly inhibit the recruitment of TRAFs to the receptors. It is more likely that the compounds interact with other members of the signalling complexes involved in RANKL and TNF signalling, leading to destabilisation of these complexes. One possible target that is common to both TNF- and RANK-signalling is the kinase TAK1, which is important for activation of both the NFκB and MAPK pathways. However, in the RANK signalling complex, TAK1 is recruited through TAB1 and TAB2,19 whereas in TNF signalling TAK1 is recruited by RIP120 and TAB1 and TAB2 are not necessary for signalling activity.21 Another possible target for the compounds could be ubiquitination which has been shown to be important in both TNF- and RANKL-induced signalling.22 ,23
Inflammatory diseases such as rheumatoid arthritis are also characterised by osteoclastic bone destruction,24 and rheumatoid arthritis is associated with increased risk of fracture.25 Steroids such as prednisolone or dexamethasone are widely used to treat inflammatory disorders such as rheumatoid arthritis. However, although very potent inhibitors of inflammation, these drugs can cause steroid-induced osteoporosis,26 further increasing the fracture risk. The novel ABD compounds described here, in contrast, couple anti-inflammatory effects with bone protective effects.
Neutralising antibodies to TNF or decoy TNF-receptors have been shown to be highly effective treatments for inflammatory diseases such as rheumatoid arthritis,27 psoriatic arthritis28 and ankylosing spondylitis,29 ,30 and there is evidence that anti-TNF therapy slows the rate of bone erosions and systemic bone loss in rheumatoid arthritis.24 ,31 ,32 However, a recent review of the available data suggests that the antiresorptive effects of anti-TNF therapy are related to control of disease activity rather than direct suppression of osteoclast activity.33 Furthermore, a study by Chopin et al34 showed that although anti-TNF therapy suppressed bone resorption markers in the early phases of the treatment, bone resorption levels had returned to baseline by 1 year of treatment. A study by Vis et al has shown that although anti-TNF treatment halts further bone loss in the spine and hip, bone loss in the hand does progress35 and a recent study of fracture risk in patients with autoimmune disease showed no decrease in fracture risk in patients treated with anti-TNF36 compared with those on conventional therapy. Preclinical studies have also shown that blockade of RANKL-induced RANK signalling is required for full protection against inflammation-induced bone destruction in vivo.3 ,6 ,37 ,38 In the present study, the antiresorptive effects of the ABD compounds could be due either to suppression of the inflammation or an anti-RANKL effect. Furthermore, the duration of the CIA experiments is too short to draw solid conclusions about the compounds’ long term protection against inflammatory bone loss. However, the direct effects of the ABD compounds on osteoclast formation and activity reported earlier8 ,9 ,10 suggest that these compounds may give additional protection from inflammatory bone loss.
To our knowledge, the ABD compounds are currently the only dual inhibitors of both TNF- and RANKL-induced signalling, and therefore these new compounds show promise as treatments for inflammation and bone destruction associated with chronic inflammatory conditions such as rheumatoid arthritis. As small molecules, the ABD compounds may be cost-effective alternatives to treatment with biologics such as anti-TNF. Furthermore, as one of the compounds, ABD328, showed oral activity, the administration of these compounds may avoid the problems and costs associated with compounds administered by injection or intravenous drip.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figures
Footnotes
Handling editor Tore K Kvien
Contributors EC performed experiments and analysed data. IRG designed experiments, designed and synthesised the novel compounds and co-wrote the paper. PM provided essential materials and advice. LR performed the experiments. MG designed and performed experiments. SHR designed experiments and co-wrote the paper. RJvH designed and performed experiments, analysed data and wrote the paper.
Funding This work was supported in part by grants from Arthritis Research UK (grant numbers: 17362 and 18328), Scottish Enterprise and NesTech.
Competing interests SHR, IRG and RJvH are inventors on a patent for the use of the compounds as anti-inflammatory drugs, held by the University of Aberdeen.
Ethics approval All animal experiments were approved by the University of Edinburgh's ethics committee.
Provenance and peer review Not commissioned; externally peer reviewed.