Article Text

Abstract
Objectives The rs1143679 variant of ITGAM, encoding the R77H variant of CD11b (part of complement receptor 3; CR3), is among the strongest genetic susceptibility effects in human systemic lupus erythematosus (SLE). The authors aimed to demonstrate R77H function in ex-vivo human cells.
Methods Monocytes/monocyte-derived macrophages from healthy volunteers homozygous for either wild type (WT) or 77H CD11b were studied. The genotype-specific expression of CD11b, and CD11b activation using conformation-specific antibodies were measured. Genotype-specific differences in iC3b-mediated phagocytosis, adhesion to a range of ligands and the secretion of cytokines following CR3 ligation were studied. The functionality of R77H was confirmed by replicating findings in COS7 cells expressing variant-specific CD11b.
Results No genotype-specific difference in CD11b expression or in the expression of CD11b activation epitopes was observed. A 31% reduction was observed in the phagocytosis of iC3b opsonised sheep erythrocytes (sRBCiC3b) by 77H cells (p=0.003) and reduced adhesion to a range of ligands: notably a 24% reduction in adhesion to iC3b (p=0.014). In transfected COS7 cells, a 42% reduction was observed in phagocytosis by CD11b (77H)-expressing cells (p=0.004). A significant inhibition was seen in the release of Toll-like receptor 7/8-induced pro-inflammatory cytokines from WT monocytes when CR3 was pre-engaged using sRBCiC3b, but no inhibition in 77H monocytes resulting in a significant difference between genotypes (interleukin (IL)-1β p=0.030; IL-6 p=0.029; tumour necrosis factor alpha p=0.027).
Conclusions The R77H variant impairs a broad range of CR3 effector functions in human monocytes. This study discusses how perturbation of this pathway may predispose to SLE.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Complement receptor 3 (CR3)/Mac1 is a heterodimeric integrin receptor. The α-chain (CD11b), encoded by the ITGAM gene, is unique to this receptor, while the β-chain (CD18) is shared with other integrin receptors. CR3 is expressed widely and at high levels on innate immune cells, including neutrophils, monocytes and macrophages. It binds a range of ligands including iC3b, ICAM-1 and fibrinogen, and it participates in functions including phagocytosis and adhesion.1 ,2 Variation at ITGAM is one of the strongest genetic risk factors in human systemic lupus erythematosus (SLE), and it is common (minor allele frequency ≈10%) in all except east Asian populations.3,–,5 It may be a moderate risk variant for systemic sclerosis, but it is not associated with other autoimmune diseases.6 ,7
Interest has focussed on the rs1143679 variant, which encodes an arginine to histidine amino acid change at position 77 (R77H) in the beta-propeller domain of CD11b. This variant had already been recognised as an antigen in neonatal alloimmune neutropenia.8 In European populations, linkage disequilibrium between ITGAM variants leads to multiple genetic associations and difficulty pinpointing functional effects to a single variant. Trans-ancestral data support rs1143679, but without extensive resequencing and replication, attributing function to a specific variant alone remains open to interpretation.5
Direct experimental data have been conflicting. A recent study reported differences in adhesion and phagocytosis between cell lines transfected with wild-type (WT) or 77H ITGAM.9 On the other hand, the 77H genotype was not reported to influence adhesion of ex-vivo neutrophils (although iC3b was not tested).8
The strength of the ITGAM association in SLE makes this a particularly important effect to understand. It may give us an insight to key pathogenic pathways that are potentially amenable to therapeutic manipulation. The relative lack of genetic data specifically supporting rs1143679, combined with the conflicting early functional reports, means that existing studies give an inadequate picture.
Methods
Reagents
Culture media, sera and salt solutions and secondary antibodies were from Invitrogen (Life Technologies, Paisley, UK); rhM-CSF was from Peprotech (London, UK); anti-CD11b, anti-CD14 and control antibodies were from ebioscience (Hatfield, UK); polyclonal antisheep erythrocyte IgM was from Cedarlane (Burlington, Ontario, Canada). Human CD11b and CD18 in the pRK5 vector was a gift of Emmanuelle Caron, Imperial College, London. The R77H mutation was introduced using a Stratagene QuikChange site-directed mutagenesis kit (Agilent Technologies, Stockport, UK). Protein ligands were from Calbiochem, Merck-Millipore, London, UK (iC3b), R&D Systems, Abingdon, UK (ICAM-1) and Enzyme Research Laboratories Swansea, UK (fibrinogen). Human DC-SIGN was a gift of Dan Mitchell, University of Warwick.
Study participants
Study volunteers were from the TwinsUK National Institute for Health Research (NIHR) bioresource. Individuals were selected on the basis of imputed genotype but rs1143679 was checked by TaqMan assay (Applied Biosystems, Life Technologies, Paisley, UK). All volunteers were healthy with no history of autoimmune disease, recent steroid or immunosuppressant use. The study was approved by the South East London Research Ethics Committee and participants gave written informed consent. Additional volunteers were recruited at the University of Erlangen-Nuremberg, with approval from the ethics committee of the Friedrich Alexander University of Erlangen-Nuremberg.
Leucocyte preparation
Processing of heparinised blood was commenced within 1 h of collection. For flow cytometry, a leucocyte-enriched fraction was obtained by sedimentation in 3% dextran-500, before staining as outlined below. Untouched human monocytes were obtained by density gradient sedimentation (Histopaque; Sigma, Dorset, UK) and purification by negative selection (Monocyte Isolation Kit II; Miltenyi Biotec, Bisley, UK). Monocyte-derived macrophages were obtained by adherence of fresh monocytes, in serum-free medium, to glass coverslips for 1 h before being grown on in RPMI supplemented with 10% fetal bovine serum (FBS), Glutamax, pyruvate, penicillin/streptomycin, non-essential amino acids and 50 ng/ml M-CSF for 6 or 7 days at which point cells were spread and strongly adherent. All ex-vivo assays were performed on fresh paired samples, with one WT and one 77H sample collected and processed at the same time.
Cell lines
COS7 simian fibroblasts (ATCC) were grown in DMEM supplemented with 10% FBS and penicillin/streptomycin. Transient transfection with CD11b/CD18 was achieved using the Amaxa Nucleofector (Lonza, Basel, Switzerland) according to the manufacturer's protocol. No significant differences between WT and 77H cell-surface expression (assessed by flow cytometry) were seen, either in terms of the percentage of positive cells or the mean fluorescence of the positive population.
Flow cytometry
Leucocytes were resuspended in Hank's balanced salt solution with 20 mM HEPES, 1 mM calcium chloride and 1 mM magnesium chloride. Unstimulated samples were kept on ice throughout. Stimulated samples were incubated at 37°C for 5 min, with 200 nM phorbol myristate acetate (PMA; Sigma) added for 10 min before staining. Residual erythrocytes were lysed and the leucocytes fixed before analysis.
Quantitative real-time PCR
Total RNA was extracted from 2×106 cells using an RNeasy kit (Qiagen , Hilden, Germany) and complimentary DNA prepared using the SuperScript III First Strand Synthesis System (Life Technologies, Paisley, UK). cDNA quantification was carried out using ABsolute quantitative PCR SYBR Green ROX Mix (Thermo Fisher, Wallham, Massachusetts, USA) on an Applied Biosystems 7300 real-time PCR system. The relative quantification of cDNA was based on the comparative Ct method, with four housekeeping gene controls, and normalised to one randomly selected homozygous donor, using DataAssist software (Applied Biosystems). For primer details see supplementary text (available online only).
Phagocytic assay
Phagocytic assay was performed using monocyte-derived macrophages transferred to serum-free medium for 1 h. Sheep erythrocytes (TCS Biosciences, Buckingham, UK) were opsonised with rabbit antisheep erythrocyte IgM (sRBCIgM) with or without human iC3b (sRBCiC3b) according to established protocols (see supplementary text, available online only).10 Opsonised sRBC were added to the macrophages and incubated for 20 min before halting phagocytosis on ice. External, engaged sRBC were distinguished from internalised sRBC by staining as previously described (see supplementary text, available online only) and the coverslips were mounted for immunofluorescent microscopy (Zeiss Axiophot, Welwyn Garden City, UK).11 ,12 Cells were scored blind for the number of internalised sRBC (Alexa-555 labelled) and the number of bound but external sRBC (dual labelled with Alexa-488 and Alexa-555). The association index was calculated as the mean engaged (internal and external) sRBC per 100 phagocytes. The phagocytic index was calculated as the mean internalised sRBC per 100 phagocytes. The percentage phagocytosis was calculated as the mean internalised sRBC/engaged sRBC per phagocyte.12
COS7 cell phagocytosis was quantified 2 days after transfection. The protocol was identical except the phagocytic incubation was 25 min and a modified association index was calculated in which COS7 cells with no associated sRBC were not counted (even with CD11b staining it was hard to distinguish autofluorescent, but potentially untransfected, COS7 cells from those expressing low level CD11b).
Adhesion assay
Ninety-six-well Maxisorp plates (Nunc, Fisher Scientific, Loughborough, UK) were coated for 24 h with 100 μl iC3b, ICAM-1 or DC-SIGN (5 µg/ml), fibrinogen (5 mg/ml), anti-CD11b ICRF44 or human serum albumin (HSA) (10 µg/ml). Plates were blocked before use with 3% HSA for 1–2 h at room temperature. Freshly isolated cells were washed and resuspended in DMEM with 20 mM HEPES, 1 mM calcium chloride, 1 mM magnesium chloride and 1% HSA before applying to cold plates (5×105/well). Plates were incubated at 37°C for 30 min, non-adherent cell aspirated and the wells gently washed with warm PBS. Cell adhesion was quantified by 0.05% crystal violet staining and measuring 595 nm absorbance (see supplementary text, available online only).13 ,14
Cytokine assay
Freshly isolated monocytes were resuspended in RPMI supplemented with 10% FBS, Glutamax, pyruvate, penicillin/streptomycin, non-essential amino acids and added to 96-well round-bottomed plates (5×105/well). sRBCIgM or sRBCiC3b were added as appropriate (5×106/well to a total volume of 100 μl), and after 1 h the Toll-like receptor (TLR)7/8 agonist R848 (Invivogen, Toulouse, France) (or PBS) was added (2 μg/ml). Supernatants were harvested after 12–16 h and frozen at –80°C. Cytokines were quantified using a Cytometric Bead Array (BD Biosciences, Oxford, UK) according to the manufacturer's instructions.
Statistical analysis
Data are expressed as mean±SEM. Data comparisons between genotype groups were made using a linear model and Huber–White sandwich estimator/robust standard errors, accounting for layers of data clustering due to assay batches and from shared genetic/environmental factors in related twins. Within-individual comparisons (as in elements of the cytokine analysis) or when all participants were unrelated (as in the phagocytic assays) comparisons were made by paired t test.
Results
CD11b expression
We analysed CD11b cell-surface expression (using anti-CD11b antibody ICRF44) in 10 WT and 10 77H homozygous volunteers (figure 1). The neutrophils were identified by foward/side-scatter profile, and monocytes by side-scatter low/CD14 high profile. Even accounting for an outlier in the 77H group with very high CD11b expression, there was no genotype-specific difference in expression by comparison of mean fluorescent intensities (p=0.33 for neutrophils, p=0.25 for monocytes). PMA activation resulted in an increase in CD11b cell-surface expression but no difference between genotype groups (p=0.42 for neutrophils, p=0.97 for monocytes).
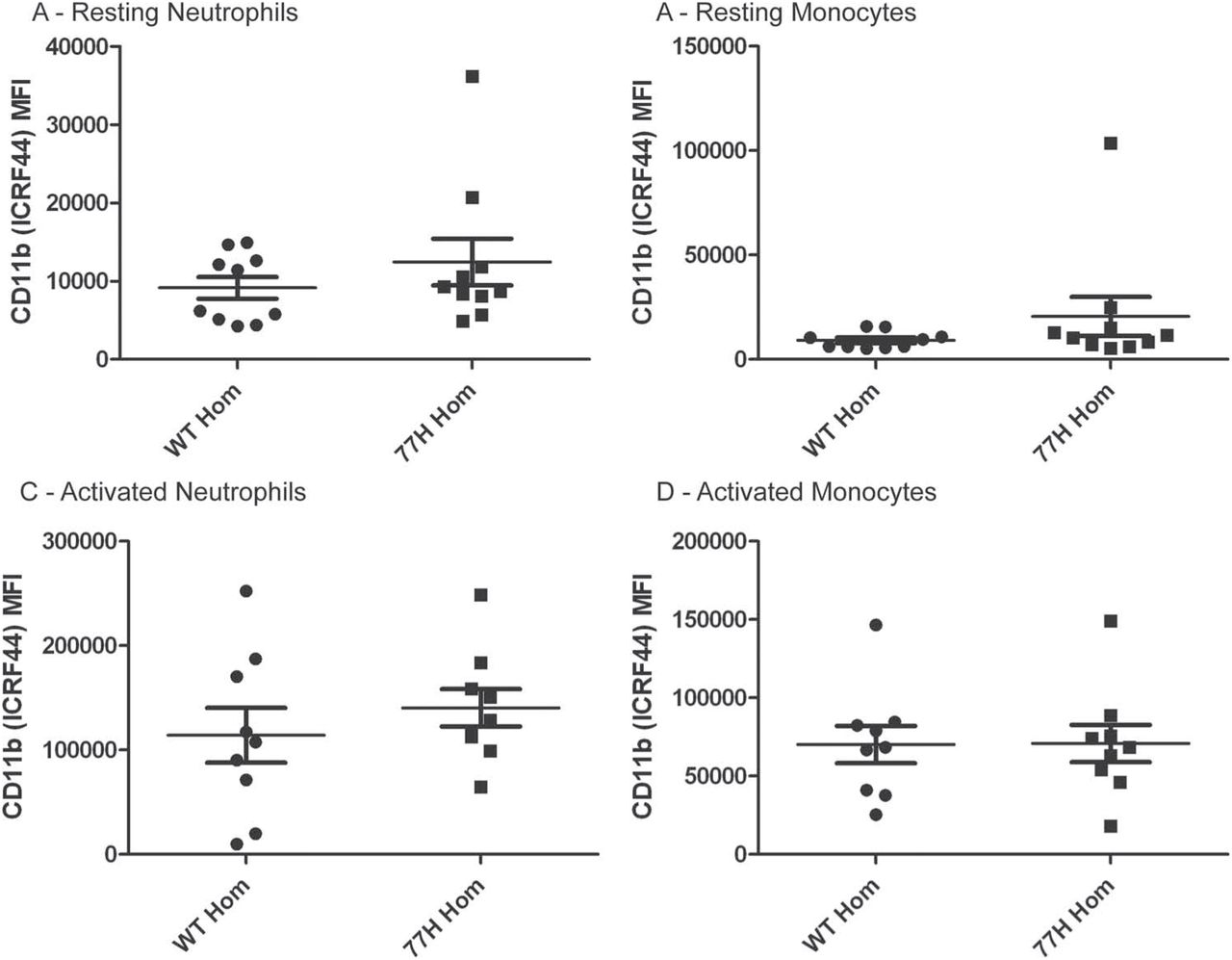
Cell-surface expression of CD11b quantified by flow cytometry. Data are presented for neutrophils and monocytes in the resting state and after 10 min stimulation with 200 nM phorbol myristate acetate. Mean fluorescence intensity (MFI) is presented. There are no significant differences between groups. ICRF, anti-CD11b antibodies.
To confirm that the rs1143679 genotype was not influencing gene expression we performed quantitative PCR in an additional 14 WT and nine WT/77H heterozygous volunteers. There was no significance difference in the relative expression of ITGAM messenger RNA (p=0.35 for unselected peripheral blood mononuclear cells, p=0.13 for monocytes; figure 2).

Relative expression of ITGAM mRNA in wild-type and heterozygous volunteers. There are no significant differences between groups. (A) Monocytes. (B) Peripheral blood mononuclear cells (PBMC).
CD11b activation/conformation
During receptor activation CD11b undergoes conformational change that can be reported using antibody CBRM1/5, which only binds the headpiece of CD11b in the active high affinity state.15 In 10 WT and 10 77H homozygous volunteers, we only observed (as expected) very low level CBRM1/5 epitope expression on unstimulated neutrophils and none on unstimulated monocytes (data not shown). PMA stimulation induced a dramatic increase in the expression of the CBRM1/5 epitope in both cell populations (figure 3), but with no genotype-specific differences (mean fluorescence: p=0.98 for neutrophils, p=0.91 for monocytes; percentage expression: p=0.49 for neutrophils, p=0.70 for monocytes).

Expression of the CD11b CBRM1/5 activation epitope on phorbol myristate acetate-stimulated monocytes and neutrophils expressed either as a mean fluorescence intensity (MFI) or a percentage of positive cells. There are no significant differences between groups.
Phagocytosis
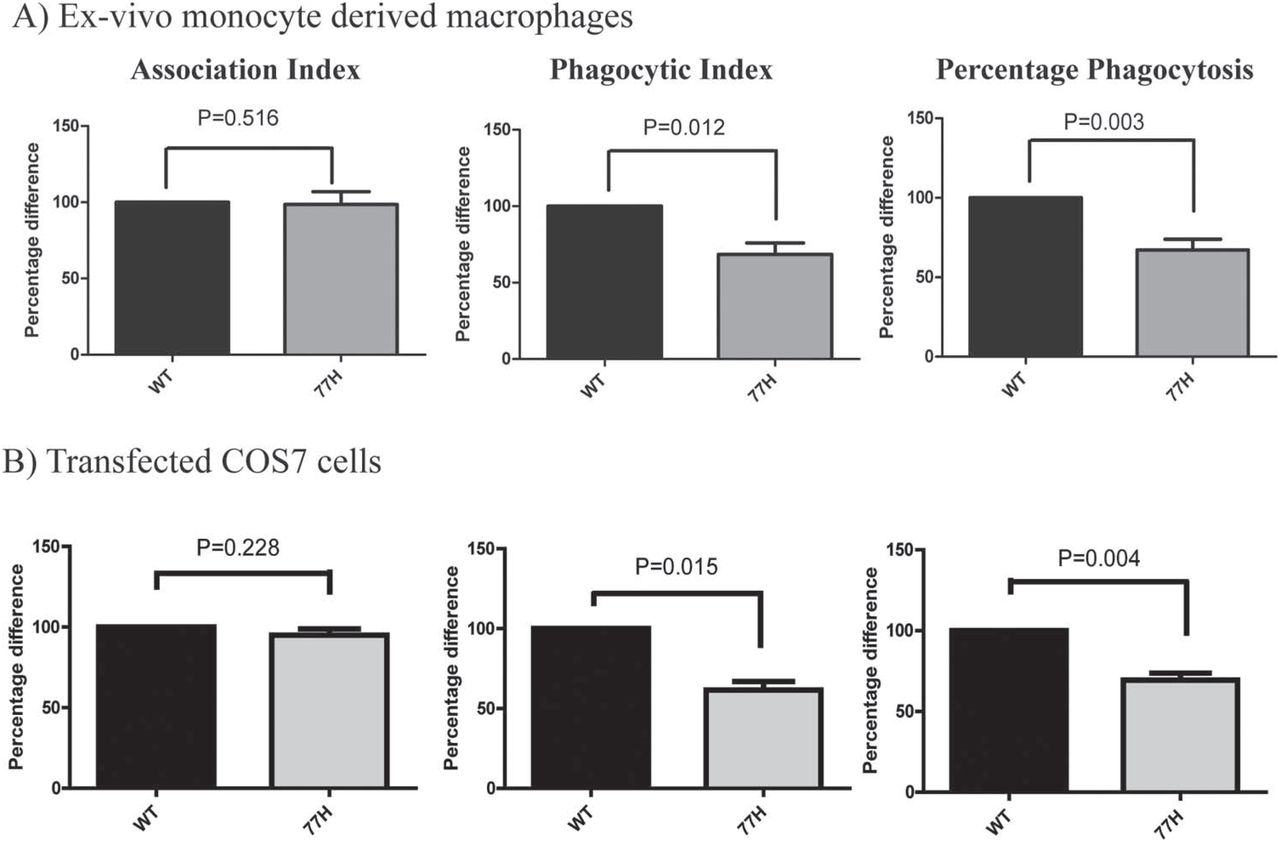
Phagocytic assay was performed using samples from seven WT and seven 77H homozygous volunteers. A mean of 258 (SD 164) macrophages per volunteer were counted, on a minimum of two coverslips. We observed no genotype-specific difference in the association of sRBCiC3b, but there was a significant reduction in phagocytosis by 77H cells quantified by phagocytic index (WT 129.3±25.1, 77H 91.6±18.8, p=0.012) or percentage phagocytosis (WT 38.9±4.6%, 77H 26.9±4.5%, p=0.003) (figure 4).

Phagocytosis of iC3b opsonised sheep erythrocytes (sRBCiC3b) by ex-vivo monocyte-derived macrophages (A) and transfected COS7 cells (B). Significant differences in phagocytic index and percentage of phagocytosis are observed, but no significant difference in association index. WT, wild-type.
To confirm that it was the 77H variant specifically that impaired phagocytosis, we replicated this assay in COS7 cells transfected with WT or 77H variant ITGAM. In five independent experiments a mean of 91 (SD 39) COS7 cells was counted from a minimum of two coverslips. Reduced phagocytosis by CD11b (77H)-expressing cells was again seen (phagocytic index: WT 90.8±14.4, 77H 53.3±6.0, p=0.015; percentage phagocytosis: WT 18.7±0.6%, 77H 12.9±0.6%, p=0.004; figure 4). No phagocytosis was seen using sRBCIgM.
Adhesion
Using 11 fresh 77H monocyte samples and 11 WT samples we observed that 77H monocytes adhered less to iC3b (595 o.d: WT 0.192±0.014, 77H 0.145±0.014, p=0.014). We also saw lower adhesion to DC-SIGN (595 o.d: WT 0.186±0.014, 77H 0.144±0.011, p=0.017), fibrinogen (595 o.d: WT 0.187±0.015, 77H 0.140±0.012, p=0.012) and ICAM-1 (595 o.d: WT 0.175±0.015, 77H 0.142±0.009, p=0.044; figure 5). We saw no difference in adhesion to plates coated with anti-CD11b ICRF44. We saw no genotype-specific difference in the binding of fresh neutrophils (resting or PMA activated) to any ligand (data not shown).

Adhesion of ex-vivo monocytes to ligand-coated plates. Adhesion of 77H monocytes expressed as a percentage of the wild-type (WT) monocyte adhesion in paired assays. Significant genotype-specific differences in the adhesion to all four CR3 ligands was observed but no difference in adhesion to anti-CD11b (ICRF) antibodies.
Cytokine secretion
We measured the secretion of interleukin (IL)-1β, IL-6, tumour necrosis factor alpha (TNFα) and IL-10 from ex-vivo monocytes from 13 WT and 13 77H homozygous volunteers. CR3 stimulation was achieved using sRBCiC3b, with sRBCIgM as control stimulation. Overnight stimulation of monocytes with sRBCIgM alone triggered a minor rise in the secretion of IL-1β, IL-6 and TNFα that was less than 1% of the level observed following TLR7/8 stimulation. Overnight stimulation with sRBCiC3b was not significantly different from that seen with sRBCIgM only.
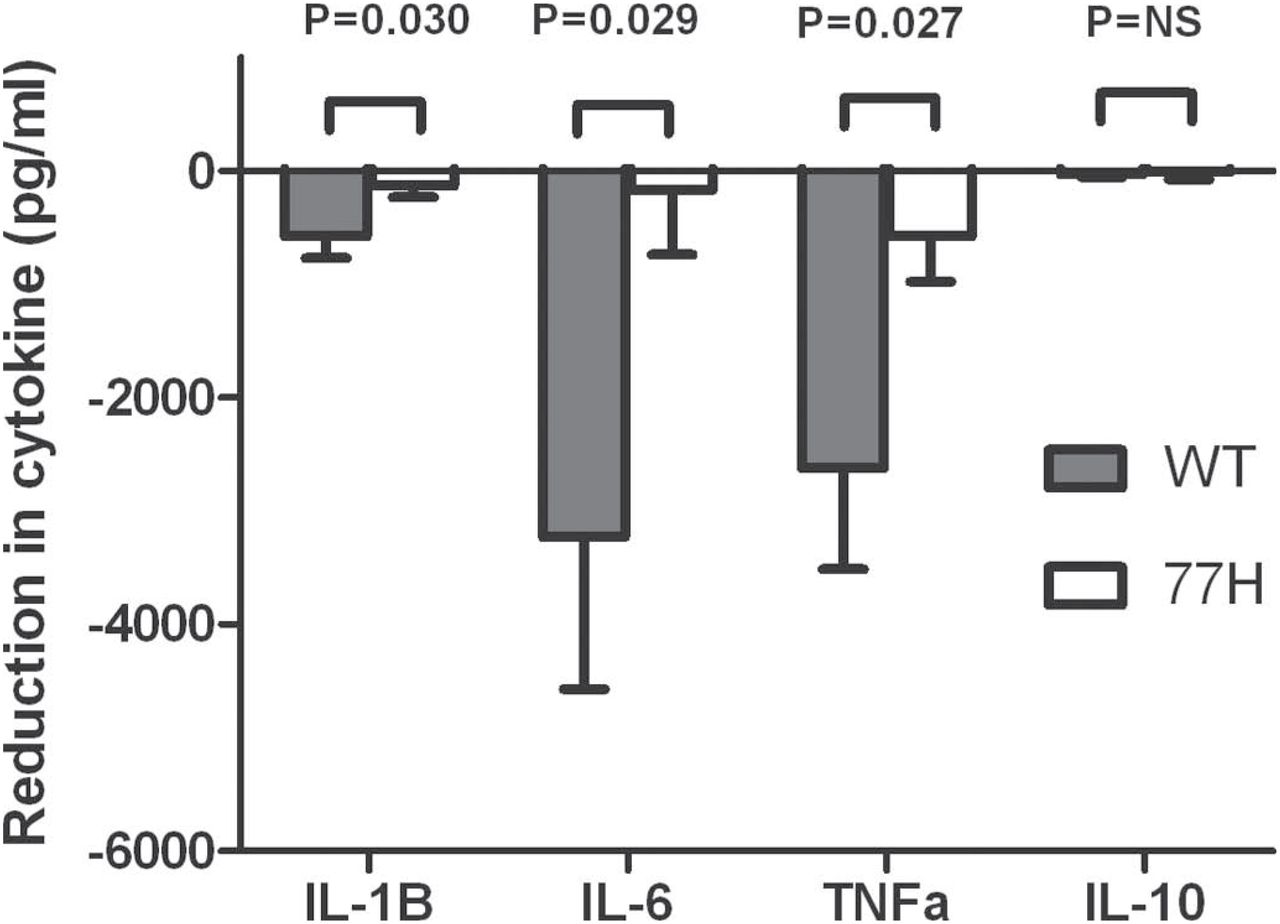
To evaluate whether CR3 signalling inhibited TLR7/8 responses we compared R848 (TLR7/8 agonist) stimulated monocytes that had been preincubated with sRBCiC3b with monocytes that were preincubated with control sRBCIgM (figure 6). The overnight secretion of pro-inflammatory cytokines by WT monocytes was significantly lower in the presence of CR3 activation (mean reduction in cytokine (sRBCIgM/R848 stimulation minus sRBCiC3b/R848 stimulation): IL-1β −577±190 pg/ml, p=0.010; IL-6 −3230±1341 pg/ml, p=0.033; TNFα −2623±891 pg/ml, p=0.012). In 77H homozygous monocytes only a modest non-significant change was seen (IL-1β −130±100 pg/ml, p=0.219; IL-6 −167±570 pg/ml, p=0.774; TNFα −557±397 pg/ml, p=0.186). A direct comparison demonstrated significant genotype-specific differences in the absolute level of cytokine inhibition (IL-1β p=0.030; IL-6 p=0.029; TNFα p=0.027). No changes in IL-10 secretion were observed in either group.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The reduction in Toll-like receptor (TLR)7/8-induced cytokine release seen when monocyte CR3 is pre-engaged using iC3b opsonised sheep erythrocytes (sRBCiC3b). The response of wild-type (WT) (shaded) and 77H cells (unshaded) is shown with p values showing the significant difference in TLR7/8 response between CD11b genotypes. IL, interleukin; TNFα, tumour necrosis factor alpha.
Discussion
We have presented an analysis of CD11b/CR3 expression and function in genotyped ex-vivo cells, demonstrating a range of impaired effector functions in cells carrying the rs1143679 polymorphism. We confirmed specific functionality of the encoded R77H variant by replicating impaired phagocytosis in genetically modified cell lines.
We observed no genotype-specific difference in the expression of CD11b by monocytes or neutrophils; an important observation when interpreting subsequent functional studies. Neither did we see genotype-specific differences in the expression of CD11b activation epitopes, even in the presence of PMA, which activates CR3 through an ‘inside-out’ signalling mechanism.16 We conclude that the R77H polymorphism does not impair CD11b inside-out signalling or the conformational changes required for receptor activation.
We observed a reduction in the phagocytosis of iC3b opsonised targets by ex-vivo monocyte-derived macrophages. This was observed despite the fact these cells may co-express complement receptor 4 (CR4), which could participate in phagocytosis and act as a source of random variability in this assay. We confirmed that impaired phagocytosis was an R77H-specific effect in variant-specific ITGAM-transfected COS7 cells, which lack phagocytic receptors but are functionally capable of phagocytosis when receptors are introduced. We also demonstrated a reduction in the adhesiveness of 77H monocytes, most notably to iC3b. Cell adhesion is not simply a function of the affinity of integrin to its ligand, but involves post-receptor-binding events that regulate receptor avidity and initiate cell spreading.17 Phagocytosis and adhesion can be considered different manifestations of the same underlying process.18 Taken together our results suggest defective ‘outside-in’ integrin signalling.
We included the dendritic cell receptor DC-SIGN as a ligand in our adhesion assay as it has been reported to bind CR3 during neutrophil–dendritic cell cross-talk.19 ,20 Our finding of a genotype-specific difference in adhesion to DC-SIGN provides additional support for an interaction between these two receptors. We observed no genotype-specific difference in neutrophil adhesion, replicating existing data.8 We suspect adhesion to alternative receptors, particularly leucocyte function antigen-1 (which binds ICAM-1 but not iC3b) accounted for the ligand-specific difference in adhesion, although the interaction between CR3 and iC3b involves more extensive binding sites than the interaction with other ligands and may lead to genuine differences in adhesion.21 ,22 The interaction between CR3 and ICAM-1 may also be assessed better under flow conditions.9 We hypothesise that because of receptor redundancy a generalised defect in adhesion due to 77H is unlikely to be an important pathogenic mechanism in SLE. SLE is not associated with pauci-cellular tissue lesions and furthermore it is difficult to reconcile an adhesion defect with the SLE specificity of the rs1143679 genetic association.6
We demonstrated genotype-specific differences in monocyte cytokine responses, with signalling through CR3 inhibiting TLR7/8 mediated pro-inflammatory cytokine release. The current literature reports that certain anti-CD11b monoclonal antibodies, or soluble mediators (particularly sCD23), induce pro-inflammatory cytokine release (including IL-1β or TNFα), either just with CR3 ligation, or as a synergistic effect with lipopolysaccharide (TLR4) activation.23 ,24 On the other hand, stimulating monocytes with sRBCiC3b has been reported to illicit a primarily anti-inflammatory response, with a downregulation of IL-12 and an upregulation of IL-10.25,–,27 This paradox is resolved by suggesting that CR3 signalling is influenced by ligand avidity: tonic, high-avidity CR3 activation producing a transient rise in pro-inflammatory cytokine production followed by prolonged inhibition of interferon-α and TLR2 and TLR4 signalling.28
The approach we (and others) take of activating CR3 through sRBCiC3b has advantages and disadvantages. The correctly orientated (thioester anchored) iC3b on sRBC is likely to trigger physiological CR3 avidity activation, but the stimulus is potentially contaminated with unconverted C3b, C1q and other factors carried over from human serum used during the opsonisation process. In addition, there is the probability of CR4 activation and perhaps activation via the controversial IgM receptor.29 Proving a CR3-specific effect through blocking experiments is difficult because anti-CD11b antibodies themselves are reported to induce cytokine release.23 ,24 Our demonstration of a genotype-specific effect actually provides, in itself, good evidence that CR3 ligation underlies the effects we observe.
Cross-talk between CR3 and TLR has also been reported elsewhere. Activation of TLR2, TLR3 and TLR4 has been associated with a greater pro-inflammatory response in itgam-/- mice and ex-vivo mouse macrophages than in WT mice or cells.30 To our knowledge we are the first to demonstrate that this inhibitory effect extends to TLR7/8. It has been proposed that one mechanism for the cross-inhibition of TLR signalling is by Syk activation with subsequent degradation of TLR-induced MyD88 and TRIF.30 As TLR7/8 also acts via the MyD88 pathway it seems intuitive that CR3 inhibits TLR7/8-induced cytokine release.
How could an under-functioning variant of CR3 increase susceptibility to SLE? Defective phagocytosis itself is clearly an important mechanism in SLE. Fc-gamma receptor gene variants (including the monocyte/macrophage genes FCGR2A and FCGR3A) that impair ligand affinity and phagocytosis are associated with SLE, while defective complement-mediated phagocytosis is also recognised as a feature of this disease.31 ,32 The ‘waste-disposal’ hypothesis proposes that impaired uptake of apoptotic cells is a key element of SLE pathogenesis, and defective uptake of apoptotic cells by lupus macrophages having been observed directly, but it is debatable whether CR3 plays a key role in this process.33,–,36 Impaired clearance of iC3b-containing immune complexes is another attractive disease mechanism, because immune complex deposition underlies much of the tissue damage in SLE.35 ,37
Our finding that CR3 ligation inhibits TLR7/8 signalling in a genotype-dependent manner is particularly interesting in the context of SLE. TLR7/8 is activated by single-stranded RNA: a component of key lupus auto-antigens such as Sm and RNP. These, in addition to synthetic TLR7/8 agonists, can activate monocytes, myeloid and plasmacytoid dendritic cells and B cells.38 ,39 It has been noted that many lupus autoantigens co-ligate both Fc receptors and TLR7/8, potentially providing pro-inflammatory signals that initiate and perpetuate autoimmunity.40 It is possible that CR3, co-ligated in the context of iC3b-containing immune complexes, provides an important immunoregulatory ‘non-danger’ signal. In the presence of the 77H polymorphism this signal may be impaired.
We deliberately adopted a ‘broad-brush’ approach to demonstrate a range of R77H effects rather than focussing on any individual mechanism in depth. We focused on monocytes because of their frequency in peripheral blood (allowing a range of assays from a limited blood source) and because fresh monocytes express relatively low levels of CR4.41 We recognise, however, that other cell types may be of great relevance in SLE, in particular CR3-expressing dendritic cell or B-cell subsets. We assume the 77H CR3 is generally under-functioning in all these cells types, but this will require experimental confirmation.
Acknowledgments
The authors acknowledge the support of Gail Clement, Karolina Zlobecka and Ayrun Nessa in assisting with the recruitment of volunteers and the technical support of Katrin Weiss.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
-
Funding The Twins UK National Institute for Health Research (NIHR) bioresource is funded by the Wellcome Trust, Europeans Community's Seventh Framework Programme (FP7/2007-2013) and ENGAGE project grant agreement (HEALTH-F4-2007-201413). The Department of Health via the NIHR comprehensive Biomedical Research Centre award to Guy's and St Thomas' NHS Foundation Trust in partnership with King's College London provided support for the Twins UK cohort and funding for flow cytometry. Original genotyping was performed by the Wellcome Trust Sanger Institute, in support of the National Eye Institute via an NIH/CIDR genotyping project. All other costs were met by an Arthritis Research UK clinician scientist fellowship (18544) awarded to BR, a fellowship within the post-doc programme of the German Academic Exchange Service (DAAD) and grant J20 of the Interdisciplinary Center for Clinical Research (IZKF) at the University Hospital of the University of Erlangen-Nuremberg awarded to BGF, and the IMI-funded project ‘Be the Cure’. TS is an NIHR senior investigator and is holder of an ERC advanced principal investigator award.
-
Ethics approval The study was approved by the South East London Research Ethics Committee. Additional volunteers were recruited at the University of Erlangen-Nuremberg, with approval from the ethics committee of the Friedrich Alexander University of Erlangen-Nuremberg.
-
Patient consent Obtained.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Correction notice This article has been corrected since it was published Online First.